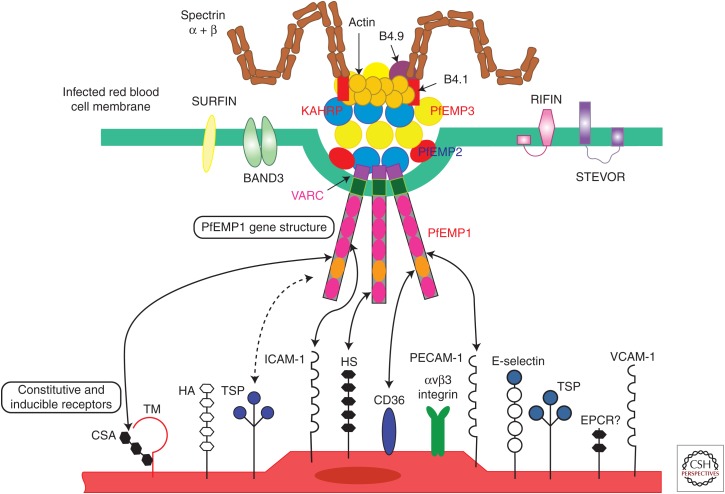
 ou aux globules rouges non parasités (rosetting) via des structures appelées « knobs » situées à la surface du globule rouge parasité. Au cours du cycle intraérythrocytaire, la prolifération et la maturation des formes parasitaires sont accompagnées par la production de protéines parasitaires (RESA, KAH-RP, MESA) qui interagissent avec des structures membranaires de l’érythrocyte (actine, spectrine) pour former des protubérances à la surface du globule rouge appelées « knobs . Ces complexes protéiques parasitaires permettent aux stades matures de P. falciparum d’échapper à la clairance splénique par séquestration dans les capillaires et veinules post-capillaires de différents organes de l’hôte mais principalement au niveau cérébral. Ce phénomène de séquestration parasitaire, lié à la cytoadhérence, a pour conséquence l’activation d’une cascade de phénomènes biologiques à l’origine de la pathogenèse de l’atteinte cérébrale de l’accès palustre grave. La cytoadhérence à l’endothélium ainsi que le rosetting réduisent la lumière des capillaires entraînant une occlusion du flux sanguin tissulaire et une hypoperfusion d’organes. L’hypoxie tissulaire et la diminution de l’élimination des produits métaboliques qui en résulte provoquent une atteinte d’organe ainsi qu’une augmentation de la lactactémie à l’origine de l’état de choc et de l’acidose métabolique. L’obstruction des capillaires et la production de toxines parasitaires (glycophosphoinositol [GPI]) induisent également une réaction inflammatoire locale liée au recrutement et à l’activation de polynucléaires neutrophiles, monocytes et plaquettes. La production par ces cellules immunitaires de médiateurs pro-inflammatoires tels que le TNF-a, l’IL-1 et l’IL-6 participe à la pathogenèse de l’accès grave via l’augmentation de l’expression de récepteurs endothéliaux impliqués dans la séquestration parasitaire et par l’altération du métabolisme du monoxyde d’azote qui joue un rôle dans l’homéostasie de la barrière hémato-encéphalique. Enfin, l’activation des cellules endothéliales après cytoadhérence parasitaire est un autre mécanisme physiopathologique de l’accès grave. L’activation des plaquettes, la libération de facteurs tissulaires endothéliales et la sécrétion de granules de Weibel-Palade contenant du facteur de von Willebrand et de l’angiotensine-2 résultent de l’activation de l’endothélium et favorisent un état procoagulant et l’altération de l’hémostase qui menace l’intégrité de la barrière endothéliale. Par conséquent, l’obstruction du flux sanguin des capillaires issue de la séquestration des formes matures parasitaires induit une hypoxie tissulaire à l’origine de l’atteinte viscérale lors d’un accès palustre grave. La réaction inflammatoire locale engendrée accompagnée par une altération de l’hémostase amplifie l’atteinte d’organes en perturbant l’homéostasie de la barrière hémato-encéphalique qui se rompt suite à l’apoptose des cellules endothéliales. Ce mécanisme physiopathologique de l’accès grave est retrouvé lors de l’atteinte cérébrale par P. falciparum. P. falciparum erythrocyte membrane protein 1 (PfEMP1) semble jouer un rôle clé dans le phénomène de cytoadhérence parasitaire et constitue donc une cible thérapeutique potentielle dans le traitement de l’accès grave [8]. Cette protéine parasitaire de 300—500 kDa, exprimée dans les « knobs » à la surface du globule rouge parasité, interagit avec des récepteurs spécifiques des cellules de l’hôte lors de la séquestration parasitaire. À l’heure actuelle, une douzaine de récepteurs de l’hôte ont été identifiés comme intervenant dans la séquestration parasitaire : héparane sulfate (HS), récepteur de complément 1 (CR1), antigène du groupe sanguin (ABO), chondroitine sulfate (CSA), PECAM/CD31, ICAM-1, CD36, thrombospondine (TSP), VCAM-1, E-sélectine, immunoglobuline non immune (Ig) et le récepteur à la protéine C (EPCR. Même si l’ensemble de ces récepteurs ont été décrits dans l’accès grave, P. falciparum ne possèdent pas un phénotype d’adhésion pour tous ces récepteurs et, par conséquent, ils n’ont pas tous la même importance en fonction de la présentation clinique et biologique de l’accès palustre. Alors que les récepteurs HS et CSA ont plus particulièrement été décrits dans le paludisme gestationnel, ABO, CR1 et Ig semblent impliqués dans le phénomène de rosetting. CD36 est un récepteur constitutif de l’endothélium dont le rôle est controvers. ICAM-1 et plus récemment EPCR sont actuellement, d’après les résultats de récentes études in vitro, les récepteurs potentiellement impliqués dans l’accès grave et plus particulièrement dans l’atteinte cérébrale chez l’enfant de moins de 5 ans en Afrique subsaharienne. L’identification du récepteur impliqué dans la survenue d’un accès grave permettrait de développer de nouvelles thérapeutiques contre la séquestration parasitaire.Dans le génome de P. falciparum, la protéine PfEMP1 est codée par une soixantaine de gènes appartenant à la famille des gènes var. Ce groupe de gènes, principalement situé dans les régions subtélomériques du chromosome, est caractérisé par une fréquence élevée de recombinaison génique à l’origine d’une grande diversification du répertoire des gènes var. La conséquence directe de ce polymorphisme génique est une grande variation antigénique de PfEMP1 qui permet au parasite d’échapper au système immunitaire de l’hôte et d’avoir un grand nombre de phéno type d’adhésion associé à la séquestration parasitaire. Cette variation antigénique et phénotypique de PfEMP1 est associée à la composition variable du domaine d’adhésion de la protéine en N-terminal composé d’une succession de domaines protéiques tels que les domaines duffy bindinglike (DBL) et les régions inter-domaine riche en cystéine (CIDR). De par leur localisation génomique, leur sens de transcription et leur nombre, les gènes var ont été classés en différents groupes : A, B, C, B/A, B/C, var1, var2csa, var3 et var4. Des études de terrain en zone d’endémie et des études in vitro ont mis en évidence l’implication des gènes var du groupe A, B et B/A dans le neuropaludisme en zone d’endémie. Dans différents génomes de souches de P. falciparum, des études d’alignement de 399 séquences protéiques de PfEMP1 des différents groupes cités précédemment ont mis en évidence 23 domaines protéiques conservées appelés domaine cassette (DC) dont certains comme DC8, DC13, DC4 et DC5 sont devenus des candidats potentiels pour l’élaboration d’un vaccin [9,15—20] | Infectiologie | Medicalis Encyclopædia){kind=link}
La pathogenèse du paludisme a un contexte large et étroit selon le cadre de référence. Dans le cycle de vie moustique-humain, les six espèces de Plasmodium, l’agent causal du paludisme infectant les humains sont Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale wallickeri, Plasmodium ovale curtisi, Plasmodium malariae et Plasmodium knowlesi. Ces espèces subissent 10 états morphologiques ou plus, se répliquent d’un à plus de 10 000 cellules, et varient dans la population totale d’un à plusieurs plus de 106 organismes. Chez l’hôte humain, seul un petit nombre de ces stades morphologiques conduisent à la maladie clinique et la grande majorité de tous les patients infectés par le paludisme dans le monde produisent peu de symptômes (voire aucun) chez l’homme. La maladie clinique humaine (par exemple, la fièvre, l’anémie, le coma) est le résultat de la biologie préprogrammée du parasite de concert avec la réponse physiopathologique humaine. Les mises en garde et les corollaires qui ajoutent des variations à cette interaction hôte-parasite comprennent la diversité génétique des parasites des protéines clés, les co-infections, les comorbidités, les retards de traitement, les polymorphismes humains et les déterminants environnementaux. Une solide compréhension de la pathogenèse du paludisme dans tous les groupes des patients est nécessaire non seulement pour prédire où la maladie (en particulier les maladies graves) se produira, mais aussi pour pouvoir la prévenir. Cet article revient sur le cycle de développement du plasmodium et présente l’essentiel des mécanismes physiopathologiques du paludisme dans ses formes simple et grave.
Citer cet article : Sophia, Les éditeurs de l’Encyclopédie. « Physiopathologie du paludisme ». Sophia Encyclopædia, https://larepublica.cd/sophiaencylopaedia/24257/, (Date d’accès).
Introduction
La biologie des parasites du paludisme Plasmodium falciparum, telle que mesurée in vitro, est finie, prévisible et facilement perturbée expérimentalement au cours du cycle de vie de 48 heures. Dans le cycle de vie moustique-humain, cependant, ce parasite, ainsi que les cinq autres espèces infectant les humains (Plasmodium vivax, Plasmodium ovale wallickeri, Plasmodium ovale curtisi, Plasmodium malariae et Plasmodium knowlesi), subit 10 états morphologiques ou plus, se réplique de 1 à plus de 10 000 cellules, et varie dans la population totale d’un à plus de 106 organismes. De plus, tous ces parasites (à l’exception de P. knowlesi chez l’homme) ont été exposés pendant des milliers de millénaires aux barrières physiques, immunologiques et plus récemment chimiothérapeutiques chez les moustiques et les humains, ce qui exerce une énorme pression de sélection sur l’espèce. Il s’agit clairement d’un programme finement réglé, bien répété et habilement exécuté. Une pression de sélection similaire a été exercée sur les humains et a abouti à des résultats évolutifs aussi fascinants que la drépanocytose, les hémoglobinopathies, les mutations de cytokines et le déficit enzymatique, qui confèrent, en tant que groupe conceptuel, la capacité de survivre jusqu’à la maturité et la reproduction.
Étiopathogénie du paludisme
Le paludisme non compliqué est défini comme la présence de symptômes (fièvre) mais aucun signe clinique ou de laboratoire indiquant la gravité ou le dysfonctionnement des organes vitaux (OMS 2015). La maladie des patients représente, cependant, un sous-ensemble de tous les individus qui ont été piqués par des moustiques infectés et une partie beaucoup plus importante de la population «à risque» présenterait un frottis de paludisme positif ou un autre test de diagnostic s’ils étaient dépistés (infection asymptomatique, vrai nombre variable et difficile à estimer). La biologie exacte du parasite du paludisme au sein de ces deux groupes est probablement très similaire, les différences essentielles étant dues à la réponse immunitaire humaine, au nombre d’infections antérieures et au profil d’exposition.
Les humains acquièrent le parasite Plasmodium via la piqûre d’un moustique anophèle infecté. Après le stade hépatique pré-érythrocytaire, les parasites plasmodium envahissent les globules rouges (GR) et commencent à circuler dans la circulation sanguine. À la différence des autres espèces, les parasites P. falciparum expriment le récepteur PfEMP1 de Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) sur les membranes des globules rouges infectés.
Les symptômes de l’infection paludéenne (tableau infectieux non spécifique d’aspect pseudogrippal caractérisé par de la fièvre, des frissons et des douleurs abdominales) ne peuvent commencer chez tout patient malade qu’avec la première rupture du schizonte hépatique et la libération de mérozoïtes dans la circulation périphérique – cet événement est silencieux pour la grande majorité des patients qui deviendront cliniquement malades. Au fur et à mesure que les parasites poursuivent leur cycle de vie asexué de réinvasion de mérozoïtes, de développement de trophozoïtes et de rupture de schizonte sur 24 à 48 heures, le niveau de parasitémie est parallèle au niveau de réponse humaine (c’est-à-dire fièvre, protéine C-réactive [CRP] et le facteur de nécrose tumoral α [TNF-α]) jusqu’à ce que le patient franchisse un seuil de conscience et «se sente malade». Chez l’hôte humain lors d’une infection initiale, l’ingestion de macrophages de mérozoïtes, de schizontes rompus ou de trophozoïtes présentateurs d’antigènes dans la circulation ou la rate entraîne la libération de TNF-α. La molécule, avec d’autres en cascade, est responsable de la fièvre lors de l’infection. D’autres molécules importantes trouvées lors d’une infection active comprennent l’interleukine 10 (IL-10) et l’interféron γ (IFN-γ) entre autres.
Dans les infections ultérieures, un certain degré de production d’anticorps produit par l’axe antérieur macrophage-cellules T-cellules B du système immunitaire confère une activité macrophage supplémentaire conduisant à une élimination plus efficace des parasites et à la production de nouveaux anticorps. Au fur et à mesure que le système immunitaire de l’hôte humain se fraye un chemin à travers le répertoire de protéines parasitaires présenté en continu, des anticorps supplémentaires se développent, conférant une protection supplémentaire.
Plasmodium falciparum
Plasmodium falciparum (Pf) modifie la surface du globule rouge infecté et crée un phénotype adhésif, qui élimine le parasite de la circulation pendant près de la moitié du cycle de vie asexué, une période unique parmi les parasites du paludisme. La liaison des érythrocytes infectés peut se produire avec l’endothélium, les plaquettes ou les globules rouges non infectés. Le parasite accomplit cet état cytoadhérent («cellule collante») par l’intermédiaire de la protéine membranaire érythrocytaire 1 de Plasmodium falciparum (PfEMP1), qui est le produit de la transcription du gène var.
Au sein d’un parasite Plasmodium falciparum donné, il existe environ 60 copies du gène var, chacune très variable et différente des autres. Ces gènes représentent certains des plus diversifiés du génome du parasite et de la population totale de parasites. Leur expression est régie par plusieurs mécanismes, notamment la pression de sélection immunitaire et l’épigénétique. Cet aspect de la biologie des parasites (expression du gène var) se produit dans toutes les infections, y compris le paludisme asymptomatique et non compliqué. Le potentiel de cette interaction homme-parasite à provoquer des maladies chez l’homme a un spectre défini discuté ci-dessous.
Indépendamment de la variabilité de la maladie, la séquestration (élimination temporaire du parasite de la circulation par la liaison à la surface des globules rouges) de Plasmodium falciparum se produit lors de chaque infection humaine pendant la moitié du cycle de vie asexuée. Ainsi, dans une infection de faible niveau dans laquelle une seule piqûre de moustique a introduit une seule couvée de parasites synchrones, les patients peuvent présenter des frottis sanguins périphériques négatifs. Cela peut être particulièrement vrai chez les voyageurs ou les résidents des régions à faible endémicité. Dans les milieux fortement endémiques, cependant, les patients sont mordus à plusieurs reprises et peuvent présenter une fièvre continue et un frottis sanguin constamment positif pendant les premières décennies de la vie. Au fur et à mesure qu’une immunité locale à la population Plasmodium falciparum évolue chez un hôte donné, les frottis peuvent à nouveau tomber à des niveaux très bas et même devenir indétectables malgré une transmission continue.
Plasmodium vivax
Plasmodium vivax (Pv) est le parasite du paludisme le plus courant causant des maladies cliniques en dehors de l’Afrique. Contrairement à Plasmodium falciparum, mais comme tous les autres parasites du paludisme humain, Plasmodium vivax ne présente pas de période prolongée de séquestration pendant l’infection. Le parasite est donc probablement plus fréquemment exposé à la clairance par la rate et plus fréquemment observé sur un frottis sanguin périphérique lors d’une infection. L’une des caractéristiques uniques de Plasmodium vivax est la préférence des globules rouges pour les réticulocytes et l’utilisation prédominante de l’antigène Duffy pour l’invasion, mais pas absolument.
Cela conduit à une infection clinique avec un niveau de parasitémie inférieur à celui observé chez Plasmodium falciparum. Comme les réticulocytes sont plus gros que les globules rouges matures, les cellules infectées apparaissent plus grosses que les cellules qui les entourent sur le frottis sanguin périphérique. Les points de Schuffner caractéristiques, qui sont des structures cavéoles–vésicules, sont observés à la fois chez Plasmodium vivax et Plasmodium ovale. La forme diagnostique de Plasmodium vivax est la forme amiboïde où le cytoplasme, unique à Plasmodium vivax, a des projections en forme de doigts sans une structure ronde à ovale typique.
Cliniquement, les patients se présentent presque de la même manière que les autres infections paludéennes avec de la fièvre et une constellation d’autres symptômes possibles. Contrairement à Plasmodium falciparum et Plasmodium malariae (qui ont une seule rupture de schizonte hépatique même peu de temps après l’invasion de sporozoïtes), Plasmodium vivax et Po peuvent «réapparaître» lorsque des hypnozoïtes (formes quiescentes qui durent des mois à des années dans le foie après une seule exposition à un sporozoïte) libèrent des mérozoïtes. Ainsi, le moment clinique d’une maladie (plusieurs mois ou années après l’exposition) pourrait être un indice de l’un de ces organismes.
Plasmodium ovale
Plasmodium ovale (Po) s’est avéré être deux espèces distinctes (Plasmodium ovale curtisi et Plasmodium ovale wallikeri), qui ne diffèrent que par une période de latence plus courte chez Plasmodium ovale wallikeri et des différences de séquence génétique. Ainsi, ces deux organismes sympatriques sont impossibles à distinguer, présentent le même syndrome clinique et répondent au même traitement. Bien que leur comportement soit similaire à Plasmodium vivax, Plasmodium ovale n’a pas besoin de l’antigène du groupe sanguin Duffy pour l’invasion des globules rouges. Sur le frottis sanguin périphérique, les formes diagnostiques de Plasmodium ovale sont la forme cométaire du trophozoïte ainsi que l’aspect ovale des globules rouges infectés et la présence de fimbria ou de projets en forme de doigts de la membrane des globules rouges. Les stades anneau, schizonte et gamétocyte de Plasmodium ovale sont très similaires à Plasmodium vivax.
Plasmodium malariae
Plasmodium malariae (Pm) est la forme la plus bénigne d’infection palustre avec plusieurs caractéristiques cliniques distinctes. Les patients ont de la fièvre toutes les 72 heures pendant une infection en raison du cycle de vie plus long du parasite. Le nombre de mérozoïtes produits à chaque rupture de schizonte est plus faible et, par conséquent, les parasitémies sont globalement plus faibles chez ces patients par rapport aux autres types de paludisme. Ce long cycle de vie et ce faible niveau d’infection conduisent à une réponse immunitaire plus robuste. Ainsi, Pm est souvent considérée comme la cause d’un paludisme chronique qui peut durer des décennies. Un résultat unique de Pm est le dépôt de complexes immuns dans les reins qui peuvent entraîner une néphrite. Sur frottis sanguin périphérique, le parasite présente la forme classique et diagnostique en «bande» ainsi qu’un schizonte avec peu de mérozoïtes et un globule pigmentaire central (de couleur dorée) appelé forme «marguerite». Cliniquement, les patients qui présentent des symptômes de paludisme et présentent des formes évocatrices de Pm doivent être évalués pour P. knowlesi ainsi que Plamodium falciparum car la détection des symptômes et/ou la probabilité de co-infection est plus élevée qu’un patient Pm véritablement symptomatique.
Plasmodium knowlesi
Plasmodium knowlesi (Pk) se trouve dans une distribution limitée en Malaisie/Indonésie Bornéo avec des cas signalés dans d’autres pays d’Asie du Sud-Est, y compris le Vietnam, Singapour, le Myanmar, le Cambodge, la Thaïlande et les Philippines. L’exposition aux moustiques qui se nourrissent de macaques à longue queue et/ou à queue de cochon est nécessaire pour la transmission, car aucune transmission interhumaine (via les moustiques) n’a été signalée. Des travaux in vitro ont montré que les parasites préfèrent les jeunes globules rouges mais peuvent, avec le temps, s’adapter pour infecter les globules rouges humains plus âgés, un phénomène qui limite actuellement la propagation rapide de l’infection au-delà du milieu humain:singe.
La maladie se présente comme les autres paludismes avec de la fièvre/des frissons et des maux de tête avec des caractéristiques peu communes comme des nausées/vomissements, des myalgies/arthralgies, des symptômes des voies respiratoires supérieures et une jaunisse (ictère). Bien que rares, des complications mortelles de Plasmodium knowlesi se sont produites et le font avec une fréquence plus élevée que celle observée chez Plasmodium vivax et Plasmodium falciparum proportionnellement en raison de la nouvelle émergence chez l’homme (zoonose) et de l’absence de temps suffisant pour l’adaptabilité humaine. Bien que Plasmodium knowlesi ne soit pas unique parmi les paludismes de vertébrés non humains qui ont été transmis à l’homme, l’émergence actuelle d’une large distribution dans la population d’une maladie à mortalité élevée n’a pas été signalée auparavant et mérite une attention particulière.
Physiopathologie du paludisme grave
Définition et critères de gravité du paludisme
Le paludisme non compliqué est facilement traité au cours de chaque épisode symptomatique avec des antipaludiques spécifiques au parasite et la grande majorité des patients éliminent facilement l’infection lorsqu’ils sont traités avec une observance appropriée. Néanmoins, dans certaines populations avec des facteurs de risque associés (âge extrême, malnutrition, immuno-dépression) ou considérées comme non immunes vis-à-vis du parasite (enfants < 5 ans, femme enceinte et voyageurs), l’infection par Plasmodium falciparum (parfois aussi les infections à Plasmodium vivax et Plasmodium knowlesi) peut conduire à un accès palustre grave défini par un ensemble de critères cliniques et/ou biologiques qui représentent à la fois des facteurs pronostiques et d’urgences pour la prise en charge du patient. Ce sont (OMS) :
⊛ Critères cliniques :
- Défaillance neurologique (Obnubilation, confusion, somnolence, coma – score de Glasgow < 11).
- Défaillance respiratoire (Sans ventilation mécanique : PaO2 < 60 mmHg ou SpO2 < 90 % en air ambiant ou fréquence respiratoire > 32cycles/min avec ventilation mécanique ou ventilation non invasive : PaO2/FiO2 < 300 mmHg). L’œdème pulmonaire est multifactoriel : surcharge, œdème lésionnel spécifique au Plasmodium, pneumonie d’inhalation.
- Défaillance circulatoire (Pression artérielle systolique < 80 mmHg avec signes périphériques, Drogues vaso-actives, Signes périphériques d’insuffisance circulatoire),
- Convulsions répétées (Au moins 2 par 24h).
- Hémorragie (Definition purement clinique), Ictère clinique.
⊛ Critères biologiques :
- Hémoglobinurie (urines rouge foncé, hémoglobinurie à la bandelette – microscopique). L’hémoglobinurie est rare. Une hémoglobinurie importante doit faire envisager un autre diagnostic : la fièvre bilieuse hémoglobinurique.
- Insuffisance rénale (créatinémie > 265µmol/l ou urée sanguine > 17 mmol/l et diurèse < 400 ml/24h (oligurie) malgré réhydratation). L’insuffisance rénale aiguë est souvent d’abord fonctionnelle, par déshydratation. La nécrose tubulaire peut être isolée ou, beaucoup plus rarement, s’inscrire dans le cadre d’une défaillance multiviscérale.
- Hypoglycémie (Glycémie < 2,2 mmol/l). L’hypoglycémie est souvent favorisée par la quinine. Le risque est majoré durant la grossesse.
- Anémie sévère (Hémoglobine < 7 g/dl, hématocrite < 20%). À la différence de l’enfant, l’anémie initiale est rare chez l’adulte non immun, sans anémie préexistante. L’hémoglobine ne diminue franchement (suite à l’hémolyse, et à l’insuffisance rénale) que dans les jours suivant le début de l’accès grave.
- Acidose (bicarbonates < 15 mmol/l ± pH < 7,35).
- Hyperlactatémie (Toute valeur > normale, lactates plasmatiques > 5 mmol/l). L’insuffisance circulatoire et l’hyperlactatémie se voient dans les formes les plus graves. Elles sont liées au paludisme lui-même et parfois à une infection bactérienne associée (pneumonie ou septicémie par translocation bactérienne digestive).
- Hyperparasitémie (> 4%, notamment chez le non-immun). Une parasitémie élevée a une valeur pronostique quand elle est associée aux autres critères majeurs. Seule, elle ne fait que traduire le retard thérapeutique.
- Ictère (bilirubinémie totale>50 µmol/l).
Les critères les plus fortement associés au pronostic sont le coma, l’acidose métabolique, l’état de choc, l’œdème pulmonaire et, à un moindre degré, l’insuffisance rénale.
Lors d’une infection à Plasmodium vivax uniquement et sans autres comorbidités, la mort due à la maladie est extrêmement rare. Cependant, en présence de comorbidités, une maladie grave et des issues fatales sont signalées. En raison du phénotype récurrent du foie, les maladies chroniques peuvent entraîner une anémie et une malnutrition sévères, qui prédisposent aux co-infections et à une faible réponse immunitaire. Comme les Plamodium falciparum et Plasmodium knowlesi graves (et toute infection grave), la voie commune finale peut inclure la détresse respiratoire, l’insuffisance hépatorénale et le choc. Le coma a été rarement signalé dans l’infection à Plasmodium vivax, mais la cause de ce coma n’est pas la même que celle observée à Plamodium falciparum (dans laquelle une séquestration parasitaire à un niveau élevé dans le cerveau est observée dans les cas mortels).
Le taux de maladie grave chez Plasmodium knowlesi est plus élevé (∼8 %), proportionnellement, que chez Plasmodium falciparum ou Plasmodium vivax et a une mortalité plus élevée (3 %). Semblable au paludisme à Plasmodium falciparum sévère chez les adultes, la maladie sévère à Plasmodium knowlesi se présente généralement avec la même constellation initiale de fièvre, etc., et progresse dans la maladie grave pour inclure l’hypotension, la détresse respiratoire, l’insuffisance rénale aiguë, l’hyperbilirubinémie et le choc. Le coma, tel qu’il est requis pour un diagnostic de paludisme cérébral à Plasmodium falciparum, n’est pas toujours observé dans les cas mortels Plasmodium knowlesi. La «voie commune» de toute infection grave (observée dans celle à Plasmodium falciparum, la septicémie bactérienne, etc.) est le résultat d’une réponse immunitaire humaine exagérée en présence d’une infection non traitée ou retardée dans le traitement et probablement pas le résultat d’une mécanismes spécifiques des organismes.
En fait, Plasmodium knowlesi en tant que cause d’autres conditions morbides (septicémie à Gram négatif) a été signalée. Pathologiquement, là où Plasmodium falciparum montre une séquestration intense dans le cerveau avec une congestion et éventuellement un gonflement du cerveau, Plasmodium knowlesi a une apparence similaire dans les tissus avec un curieux manque d’ICAM-1 dans le cerveau. La famille Plasmodium knowlesi de gènes homologues aux gènes var de Plasmodium falciparum sont les SICAvars, qui sont plus grands en structure (12 contre deux exons) et en quantité (>200 contre 60) que les gènes var de Plasmodium falciparum. Le mécanisme exact et les interactions de Plasmodium knowlesi avec l’endothélium humain pour produire la séquestration restent à élucider.
Formes du paludisme grave et chevauchement
Chez les patients adultes, le paludisme grave a de multiples modalités (formes) en jeu dans les cas mortels, notamment des complications respiratoires, hépatiques, rénales et vasculaires.
Mais chez les enfants, le paludisme grave est souvent une présentation monosyndromique avec peu ou pas de complications manifestes. Trois formes de présentations prédominent chez les patients pédiatriques : (1) le coma (neuropaludisme ou paludisme cérébral), (2) l’anémie sévère (forme anémique), (3) et/ou la détresse respiratoire (avec acidose, ou forme respiratoire). Un chevauchement de l’Anémie sévère, de l’acidose et/ou du paludisme cérébral peut se produire, ce qui peut entraîner une mortalité plus élevée dans les groupes de chevauchement. Comme il n’existe pas actuellement de traitement auxiliaire au-delà des antipaludiques et des thérapies de soutien, la maitrise du paludisme cérébral reste un facteur clé dans le pronostic vital.
Paludisme cérébral (neuropaludisme)
La capacité unique de Plasmodium falciparum à se lier à l’endothélium (cythoadhérence) produit le syndrome clinicopathologique de paludisme cérébral. Les manifestations cliniques de paludisme cérébral peuvent commencer par une présentation typique du paludisme et rapidement (en quelques minutes à quelques heures) dégénérer en un état comateux. Après exclusion d’autres causes possibles de coma (état post-critique, hypoglycémie, méningite, septicémie bactérienne, traumatisme crânien/hémorragie cérébrale, etc.), un diagnostic clinique de paludisme cérébral peut être posé, qui est mieux confirmé par un examen de la rétine (Fond d’œil) à la recherche de signes de rétinopathie palustre (hémorragies rétiniennes, blanchiment de la rétine, décoloration des vaisseaux, un œdème papillaire, une atteinte maculaire). Dans les milieux fortement endémiques, les enfants de moins de 5 ans sont les plus exposés à la maladie avec une mortalité de 10 % à 20 %. Dans les milieux à faible endémie, tous les âges sont à risque et la mortalité peut être plus élevée chez les adultes. Dans la population non immune (les voyageurs, les militaires, etc.), un faible niveau d’infection (<1 % de parasitémie) peut entraîner des signes cliniques de paludisme cérébral et mettre la vie en danger.
Dans tous les cas, la caractéristique pathologique diagnostique de la maladie – à l’autopsie – est la présence de parasites Plasmodium falciparum dans plus de 20 % des capillaires du cerveau par frottis tissulaire ou coupes histologiques. D’autres caractéristiques pathologiques qui sont présentes de manière variable comprennent un thrombus de fibrine, les hémorragies annulaires, la décoloration du cerveau, les lésions axonales et les fuites capillaires. Les vaisseaux cérébraux apparaîtront congestionnés dans tous les cas avec un gonflement cérébral plus important dans les décès aigus tels que les patients pédiatriques africains (<48 heures). La défaillance multiviscérale et le syndrome de détresse respiratoire aiguë avec lésions alvéolaires diffuses sont plus fréquents chez les patients adultes, en particulier ceux qui ont une évolution prolongée de la maladie.
La pathobiologie du paludisme cérébral n’est pas complètement comprise, mais un grand nombre de preuves issues d’études cliniques et pathologiques ont impliqué une série d’événements et de voies à l’œuvre dans le paysage de la maladie. L’activation endothéliale vers un état plus « collant » (cythoadhérence) est la première étape. Au cours des premières phases de toute infection palustre, la stimulation des macrophages conduit à la production de TNF-α, dont l’augmentation stimule l’affichage de molécules d’adhérence dans l’endothélium cérébral comme la molécule d’adhérence intracellulaire 1 (ICAM-1).
D’autres stimulations de ce type conduisent à une variété d’autres événements de régulation à la hausse, comme résumé dans la figure 1. Un grand nombre de parasites du paludisme peuvent se lier, via PfEMP1, à des molécules omniprésentes telles que CD36 (plaquettes et endothélium à l’extérieur du cerveau), ce qui explique logiquement à la fois la thrombocytopénie de l’infection palustre ainsi que la très faible incidence du paludisme cérébral par rapport à toutes les infections palustres. Chez l’enfant africain de moins de 5 ans, la combinaison des facteurs conduisant à l’augmentation du phénotype «collant» est très probablement un retard de traitement d’une infection palustre couplé à un manque d’anticorps spécifiques protecteurs bien développés dans le cadre d’une mauvais état de santé général (par exemple, malnutrition).
Pour les personnes infectées en dehors de ce cadre, l’absence totale d’immunité entraîne la maladie. La capacité du système immunitaire humain à éliminer une maladie, qui repose presque exclusivement sur la phagocytose des macrophages, est le compartiment macrophage. Les anticorps accélèrent ce processus en augmentant l’efficacité de l’absorption. Dans les études d’autopsie d’enfants, la rate, le principal site d’élimination des parasites chez la plupart des patients à paludisme cérébral, présente une charge importante de pigments du paludisme (hémozoïne) et de macrophages, mais pas de parasites. Ceci suggère que la capacité de clairance de la rate est élevée et efficace. Là où le processus de clairance se décompose peut être dans la capacité des monocytes/macrophages circulants à suivre la séquestration et le cycle de vie du parasite (toutes les 48 heures).
Rétinopathie paludique
La pathogenèse de la rétinopathie palustre est associée à celle du paludisme cérébral, c’est-à-dire la séquestration des globules rouges infectés dans la microvascularisation rétinienne et cérébrale, provoquant une obstruction des vaisseaux et une réduction du flux sanguin entraînant une ischémie et une hypoxie en aval. La dérégulation de la voie de l’angiopoïétine-Tie-2, un régulateur important de la fonction des cellules endothéliales et de l’intégrité des vaisseaux, a été associée à la fois à la rétinopathie et à la mortalité dans le paludisme cérébral pédiatrique. De nouvelles recherches montrent qu’une expression accrue de la protéine membranaire érythrocytaire 1 de Plasmodium falciparum (PfEMP1) sur les érythrocytes de l’hôte due au parasite est corrélée à une gravité accrue du paludisme cérébral. De plus, les taux sériques de protéine 2 riche en histidine (HRP2), une protéine parasite du paludisme actuellement d’importance inconnue, peuvent être élevés chez les patients atteints de rétinopathie palustre. Une étude récente a développé un modèle rétinien chez la souris pour étudier plus avant le paludisme cérébral – en particulier, ils ont découvert que les parasites du paludisme traversent la barrière hémato-rétinienne et infiltrent potentiellement la neurorétine à travers les cellules gliales de Müller.
Les changements caractéristiques de la rétinopathie palustre consistent en un blanchiment de la rétine, une décoloration des vaisseaux, des hémorragies rétiniennes et un œdème de la papille optique. Les découvertes du fond d’œil sont généralement symétriques entre les deux yeux. Le diagnostic différentiel se fait avec : la commotion de la rétine, la rétinopathie de Purtscher, la rétinopathie de type Purtscher, l’occlusion veineuse rétinienne, et le syndrome du bébé secoué. Bien qu’un traumatisme puisse être associé à un blanchiment rétinien, à des hémorragies et à un œdème de la papille optique, il n’est pas connu que le traumatisme provoque une décoloration blanche ou orange du système vasculaire rétinien.
Thrombocytopénie et paludisme
La thrombocytopénie, définie comme une numération plaquettaire inférieure à 150×103cellules/L, est une caractéristique reconnue du paludisme à Plasmodim falciparum chez les adultes et est fréquente chez les enfants. Bien que les mécanismes qui peuvent contribuer à la thrombocytopénie ne soient pas clairs, la séquestration des plaquettes et d’autres facteurs de la microvascularisation cérébrale ont été associés à la mort chez les enfants du Malawi. Par ailleurs, il est maintenant connu que les plaquettes peuvent adhérer aux cellules endothéliales par l’intermédiaire de diverses molécules telles que le CD36. En plus de diverses cellules endothéliales et monocytes, les globules rouges humains qui expriment CD36 sont capables de se lier aux plaquettes. En tant que tel, CD36 pourrait agir comme un récepteur pour certains globules rouges parasités (parasitized red blood cells ou PRBC) et pourrait donc jouer indirectement un rôle important dans la séquestration des plaquettes et des leucocytes dans les vaisseaux sanguins du cerveau. Les plaquettes sont également connues pour jouer un rôle en facilitant la agglutination des PRBC, une caractéristique qui est également connue pour caractériser le paludisme grave.
Paludisme placentaire
Pendant la grossesse, les globules rouges infectés expriment une variante du récepteur PfEMP1 appelé récepteur VAR2CSA qui possède un domaine γ actif de type Duffy-binding, qui permet l’adhérence et la séquestration des globules rouges infectés dans les espaces intervilleux du placenta. La protéine polymorphe VAR2CSA est capable de médier l’interaction des érythrocytes infectés avec une variété de cellules hôtes, y compris les syncytiotrophoblastes placentaires et les leucocytes, mais également avec des composants du système immunitaire tels que les IgM non spécifiques. Même si une protection contre la maladie peut être progressivement acquise au fil des grossesses successives, le parasite du paludisme a développé un large panel de mécanismes d’évasion pour échapper aux mécanismes de défense de l’hôte et manipuler le système immunitaire à son avantage. On pense que le paludisme placentaire se produit via l’évitement par Plasmodium de la clairance de la rate par l’expression de la protéine VAR2CSA qui se lie au sulfate de chondroïtine A (CSA) dans l’espace intervilleux placentaire. Les protéines PfEMP1 du parasite qui sont des produits des gènes var2CSA se lient au CSA lorsque les parasites traversent le placenta, les retirant de la circulation, tandis que les parasites non liés au CSA continuent de circuler.
Le paludisme placentaire se caractérise par une accumulation massive d’érythrocytes infectés dans l’espace intervilleux du placenta, accompagnée d’une infiltration des monocytes/macrophages maternels, en particulier de monocytes. L’inflammation locale qui en résulte (placentite palustre) et l’obstruction des échanges materno-fœtaux peuvent entraîner des conséquences cliniques graves pour la mère et l’enfant. Une infiltration inflammatoire proéminente par des monocytes/macrophages provoquant une intervillosite chronique massive est liée au paludisme placentaire sévère. La réponse inflammatoire au paludisme placentaire inhibe la signalisation mTOR qui est essentielle.
L’intervillosite due au paludisme placentaire est associée à une augmentation de la formation d’autophagosomes mais à une diminution de la fusion autophagosome/lysosome entraînant une accumulation d’autophagosomes dans les syncytiotrophoblastes bloquant l’absorption placentaire des acides aminés. Chez les mères atteintes de paludisme placentaire, les gènes liés à l’autophagie sont régulés à la baisse, entraînant une dérégulation de l’autophagie et donc une altération du transport transplacentaire des acides aminés. De plus, le blocage de la signalisation mTOR dû au paludisme placentaire entraîne une diminution de l’absorption placentaire des acides aminés.
Récemment, il a été découvert que le paludisme placentaire stimule l’expression placentaire des inflammasomes qui sont liés à la sécrétion placentaire et à la maturation de l’IL-1β, une cytokine pro-inflammatoire qui provoque une diminution de l’expression du transporteur de nutriments. Ainsi, au cours du paludisme placentaire, l’environnement placentaire habituellement anti-inflammatoire évolue vers un état pro-inflammatoire. La gravité de cet état inflammatoire est un facteur important qui modifie les réponses fœtales et maternelles au cours du paludisme placentaire.
Des niveaux accrus de cytokines pro-inflammatoires, de stress oxydatif et d’apoptose entraînent des modifications pathologiques du placenta et de mauvais résultats de grossesse. Il a été démontré que les modifications histopathologiques et le paludisme placentaire augmentent le risque de prééclampsie chez les femmes enceintes, en particulier chez les primigestes.
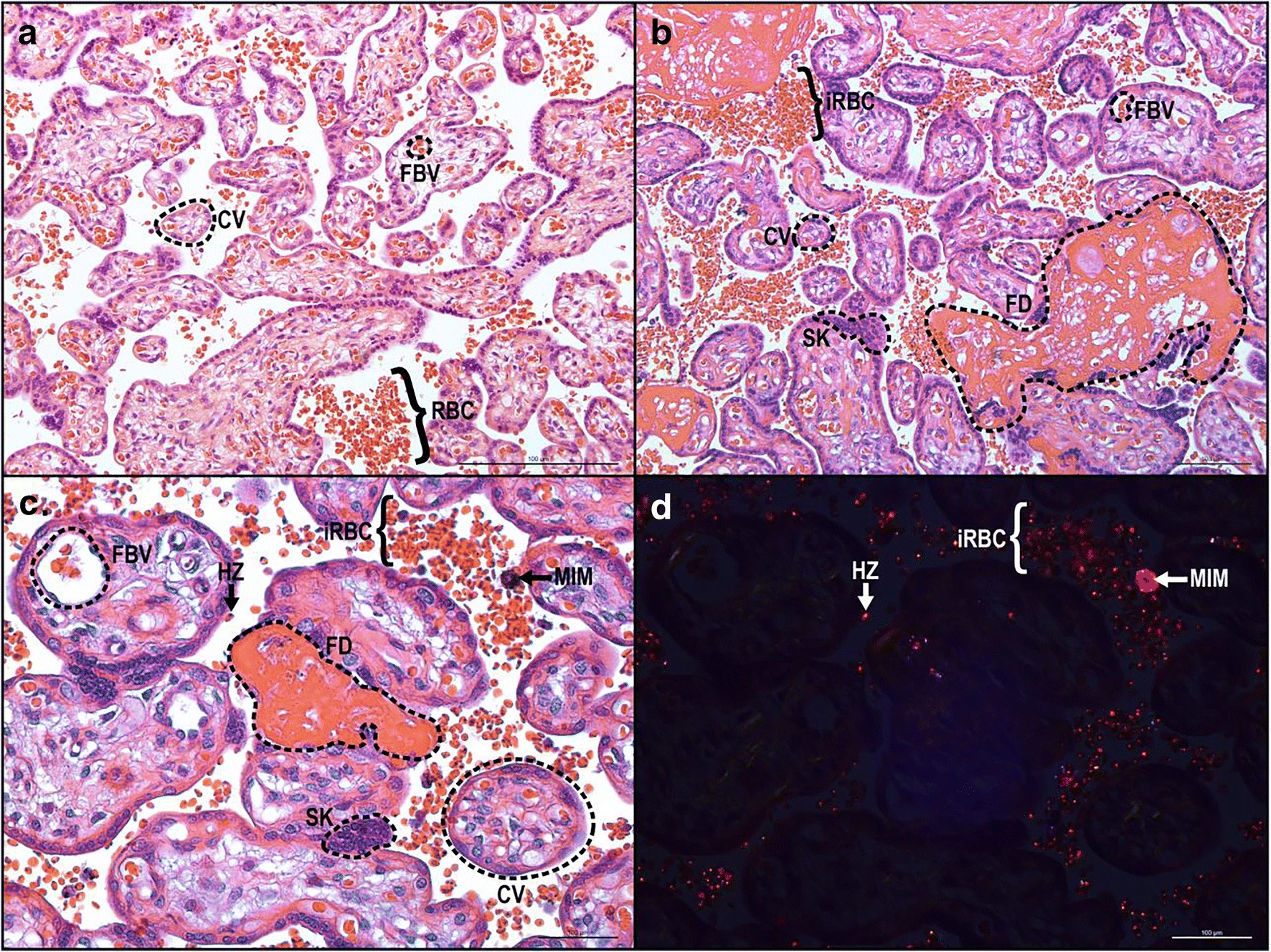
Les changements histopathologiques au cours du paludisme placentaire comprennent la présence d’hémozoïne, le dépôt périvilleux de fibrine, la formation de nœuds syncytiaux et la diminution de la surface villositaire (Figure 2).
{kind=link}
Ces altérations pathologiques du placenta peuvent limiter l’échange de nutriments entre la mère et le fœtus, augmentant le risque de retard de croissance fœtale et de bébés de faible poids à la naissance. Aussi, le paludisme placentaire diminue l’abondance de mégaline et de DAB2 dans les syncytiotrophoblastes, ce qui peut être associé à un faible poids à la naissance. L’infection palustre au début de la grossesse entraîne des altérations de la structure vasculaire du placenta, telles qu’une diminution du volume des villosités de transport et une augmentation de la distance de diffusion et de la surface des vaisseaux de diffusion, qui influencent le poids à la naissance et la durée de la gestation. Même ainsi, l’infection à Plasmodium au milieu de la grossesse a été associée à un risque accru d’accouchement prématuré, probablement en raison des changements dans les états angiogéniques, métaboliques et inflammatoires. En plus de Plamodium falciparum, il a été rapporté que Plasmodium vivax était associé à des complications pendant la grossesse, notamment l’anémie, les fausses couches, l’insuffisance pondérale à la naissance et le paludisme congénital. La pathologie placentaire chez Plasmodium vivax ne montre cependant pas le même degré d’atteinte parasitaire ou monocytaire et reste à élucider.
Anémie sévère
Les mécanismes exacts des voies qui mènent à l’anémie palustre sévère ne sont pas bien compris. La perturbation de la réponse immunitaire des monocytes et des lymphocytes en présence d’hémozoïne peut conduire à une régulation inappropriée de l’érythropoïétine (via l’IL-6, régulée à l’activation, exprimée et sécrétée par les lymphocytes T normaux [RANTES], et la protéine inflammatoire des macrophages 1s [MIP-1s]). L’élimination complète de la membrane des globules rouges et des globules rouges par la rate de manière accélérée en raison de la présence du paludisme est aussi sugerée.
Acidose et détresse respiratoire
L’acidose est un état métabolique complexe avec une gamme d’étiologies. Dans le cadre du paludisme, l’acidose est causée par une combinaison de plusieurs facteurs. Le parasite du paludisme produit la lactate déshydrogénase de Plasmodium (pLDH), qui crée de l’acide lactique entraînant une diminution du pH. La détresse respiratoire est une caractéristique courante du paludisme grave et, par séquestration, somnolence et/ou gonflement du cerveau, la suppression centrale directe des centres respiratoires entraîne des schémas respiratoires irréguliers dans le cadre de l’acidose, ce qui peut contribuer au déséquilibre du pH. Une thérapie de soutien pour protéger les voies respiratoires et rééquilibrer plus agressivement le pH peut réduire la mortalité.
Pour aller plus loin
- Zakama, A.K., Ozarslan, N. & Gaw, S.L. Placental Malaria. Curr Trop Med Rep 7, 162–171 (2020). https://doi.org/10.1007/s40475-020-00213-2
- Jamie M. O’Sullivan, James S. O’Donnell; Platelets in malaria pathogenesis. Blood 2018; 132 (12): 1222–1224. doi: https://doi.org/10.1182/blood-2018-08-865618
- G. Koki, W. Ngoulou, A.F. Nomo, S. Nguefack, E. Epee, A.L. Bella, Atteintes rétiniennes du neuro-paludisme ou «rétinopathie paludique» à Yaoundé, Journal Français d’Ophtalmologie, Volume 42, Issue 7, 2019, Pages 753-761. https://doi.org/10.1016/j.jfo.2019.03.013.
- Paquet-Durand, F., Beck, S.C., Das, S. et al. A retinal model of cerebral malaria. Sci Rep 9, 3470 (2019). https://doi.org/10.1038/s41598-019-39143-z
- Milner DA Jr. Malaria Pathogenesis. Cold Spring Harb Perspect Med. 2018 Jan 2;8(1):a025569. doi: 10.1101/cshperspect.a025569.