 : il permet d’assurer de nombreuses fonctions nécessaires au bon fonctionnement de l’organisme dont les principales sont : la production de bile pour soutenir la digestion, le maintien de l’homéostasie glucido-lipidique, ainsi que le métabolisme de molécules endogènes et des xénobiotiques. Le foie, en plus de son rôle de stockage, est également le lieu de synthèse des acides aminés et des protéines sanguines, parmi d’autres fonctions qui lui sont attribuées. L’ensemble de ces fonctions hépatiques est très finement régulé par des hormones du métabolisme, notamment l’insuline, permettant ainsi une adaptation constante de l’organisme à son environnement.){kind=link}
Le foie est un organe essentiel du corps humain. Il permet d’assurer de nombreuses fonctions nécessaires au bon fonctionnement de l’organisme dont les principales sont : la production de bile pour soutenir la digestion, le maintien de l’homéostasie glucido-lipidique, ainsi que le métabolisme de molécules endogènes et des xénobiotiques. Le foie, en plus de son rôle de stockage, est également le lieu de synthèse des acides aminés et des protéines sanguines, parmi d’autres fonctions qui lui sont attribuées. L’ensemble de ces fonctions hépatiques est très finement régulé par des hormones du métabolisme, notamment l’insuline, permettant ainsi une adaptation constante de l’organisme à son environnement.
1. Anatomie du foie
1.1. Présentation générale
Dans l’espèce humaine, le foie est l’organe le plus volumineux du corps, constituant 2 à 5% du poids du corps chez l’adulte. Asymétrique, il est situé dans la région abdominale, sur le côté droit du corps, au niveau sous phrénique (sous le diaphragme) et il se loge dans plusieurs quadrants abdominaux : l’hypochondre droit, l’épigastre et l’hypochondre gauche. Le foie est constitué de quatre lobes, le lobe droit qui se retrouve surtout au niveau de l’hypochondre droit et qui est le plus volumineux, le lobe carré et le lobe caudé que l’on ne peut voir qu’au niveau postérieure et le lobe gauche qui est séparé du lobe droit par le ligament falciforme. Au niveau sous hépatique, on retrouve la vésicule biliaire qui est accolée au foie. L’unité fonctionnelle du foie est appelée espace porte ou lobule, et possède généralement une forme hexagonale avec à chacun de ses coins une triade portale (composée d’une veine porte, une artère hépatique et un canal biliaire). Le foie contient environ 1 à 1,5 million de ces lobules hépatiques. La base des lobules (figure1) est constituée de cellules parenchymateuses, les hépatocytes, qui sont interconnectées et qui font face à des canaux sanguins appelés sinusoïdes de chaque côté. Les sinusoïdes forment des tubes enveloppés de lignes de cellules endothéliales fenêtrées. On peut trouver dans les sinusoïdes d’autres types cellulaires tels que les macrophages, connus sous le nom des cellules de Kupffer et les cellules stellaires (cellules étoilées ou cellules de Ito). Les cellules stellaires sont localisées dans l’espace de Disse, formé entre les cellules endothéliales et les hépatocytes. Du fait de leurs localisations par rapport à la triade portale, les cellules hépatocytaires sont divisées en 3 zones qui influent sur leurs fonctions physiologiques dans le foie : la région périportale, la région intermédiaire et la région péricentrale (Figure 1). Les facteurs pouvant influencer cette zonation sont multiples et peuvent comprendre l’action de gradient de morphogènes (Wnt, Hedgehog…), facteur de croissance (HGF), ainsi que d’autres facteurs tels que l’oxygène.
Figures 1 : Micro architecture du foie, gradient d’oxygène et zonation du métabolisme. 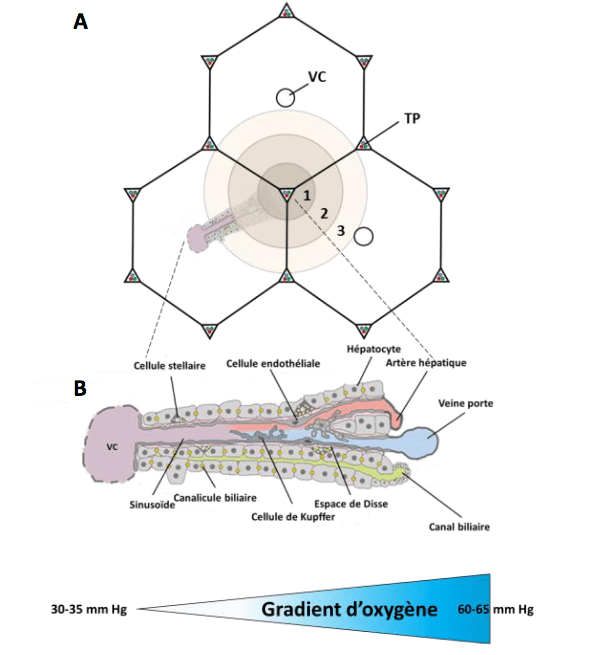 Figure 1A. (A) Lobule hépatique classique de forme hexagonale avec une veine centrale (VC) au centre et la triade porte (TP) dans chacun des coins avec une branche de la veine porte et une branche de l’artère hépatique ainsi que des voies biliaires. 3 zones peuvent être définies : 1, la zone périportale (pp) ; 2, la zone intermédiaire ; 3, la zone péri-veineuse (pv) ou centro-lobulaire. (B) Sinusoïde hépatique et gradient d’oxygène. Les hépatocytes sont connectés entre eux et produisent la bile qui est transportée par les canalicules biliaires jusqu’au canal biliaire. Les sinusoïdes sont entourées de cellules endothéliales fenêtrées. Hépatocytes et cellules endothéliales sont séparées par l’espace de Disse, qui renferme les cellules stellaires hépatiques. Les cellules de Kupffer se trouvent également dans la sinusoïde. © Adapté de (Kietzmann, 2017)
Figure 1A. (A) Lobule hépatique classique de forme hexagonale avec une veine centrale (VC) au centre et la triade porte (TP) dans chacun des coins avec une branche de la veine porte et une branche de l’artère hépatique ainsi que des voies biliaires. 3 zones peuvent être définies : 1, la zone périportale (pp) ; 2, la zone intermédiaire ; 3, la zone péri-veineuse (pv) ou centro-lobulaire. (B) Sinusoïde hépatique et gradient d’oxygène. Les hépatocytes sont connectés entre eux et produisent la bile qui est transportée par les canalicules biliaires jusqu’au canal biliaire. Les sinusoïdes sont entourées de cellules endothéliales fenêtrées. Hépatocytes et cellules endothéliales sont séparées par l’espace de Disse, qui renferme les cellules stellaires hépatiques. Les cellules de Kupffer se trouvent également dans la sinusoïde. © Adapté de (Kietzmann, 2017)
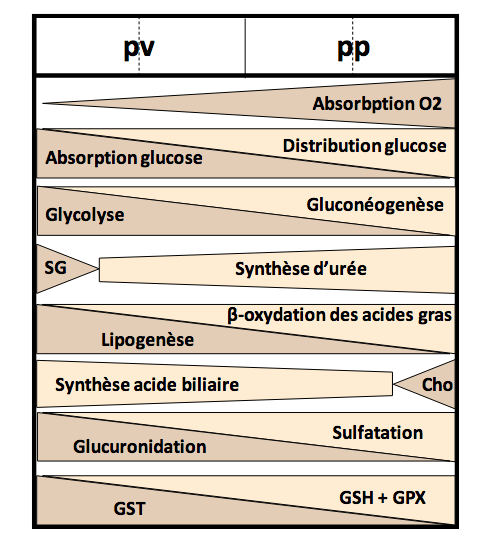 Figure 1B. La distribution des principales voies métaboliques. SG, synthèse de glutamine; Cho, synthèse de cholestérol; GST, glutathion S-transférase ; GSH, glutathion; GPX, glutathion peroxydase. © Adapté de (Kietzmann, 2017)
Figure 1B. La distribution des principales voies métaboliques. SG, synthèse de glutamine; Cho, synthèse de cholestérol; GST, glutathion S-transférase ; GSH, glutathion; GPX, glutathion peroxydase. © Adapté de (Kietzmann, 2017)
1.2. La vascularisation hépatique
Le foie est connu pour être très irrigué. Cela vient principalement de deux entités vasculaires : d’une part la veine porte qui drainant le sang veineux des intestins, supplémente le foie de matières nutritives avant de rejoindre la veine cave par l’intermédiaire des veines sus-hépatiques ; et d’autre part l’artère hépatique qui vient du tronc cœliaque (lui-même étant une branche aortique) et qui apporte au foie un sang riche en oxygène.
A. Le système porte : Le tronc porte amène au foie le sang provenant de la quasi-totalité du tube digestif ainsi que celui de la rate et du pancréas. Comme chaque partie du corps humain, le système porte est soumis à des variantes anatomiques, ce qui fait que le drainage du sang par le système porte peut-être différent d’une personne à une autre. Toutefois, la forme modale, c’est-à-dire la variante anatomique la plus retrouvée chez l’humain, correspond à la fusion de la veine mésentérique supérieure et du confluent splénomésaraïque, lui-même formé de l’union de la veine splénique et de la veine mésentérique inférieure. Il est possible de séparer le système porte en deux. En effet, on peut distinguer d’une part le système porte avant la vascularisation du foie, et d’autre part le système porte après le passage du sang au niveau hépatique. La différence ne vient pas seulement de la composition du sang veineux qui est bien évidemment différente avant et après le passage dans le foie, mais elle vient également des zones anatomiques que draine le système porte qui sont bien distinctes avant et après que le sang ait irrigué le foie.
La veine porte est une veine de calibre important venant de la combinaison entre la veine splénique, la veine mésentérique inférieure et la veine mésentérique supérieure. Plus haut, se jette trois autres veines : (1) la veine pancréatico-duodénale super-postérieure, (2) la veine gastrique droite et (3) la veine gastrique gauche. La veine porte draine donc le sang veineux venant des intestins, ce qui fait que ce sang est riche en nutriments et/ou en toxiques provenant du passage des aliments et des xénobiotiques à travers les entérocytes lors de leur ingestion. Dans le foie, la veine porte se divise en deux branches : la branche portale droite et la branche portale gauche qui elles-mêmes se subdivisent en sous branches afin de drainer le foie. Ces bifurcations se font le plus souvent au niveau extra-hépatique mais on peut avoir des bifurcations intra-hépatiques ou juste à l’entrée du foie chez certaines personnes. Ces variantes anatomiques sont à prendre en compte chez les personnes éligible à la chimioembolisation dans le cadre du carcinome hépatocellulaire pouvant survenir dans le cadre d’une cirrhose et être également responsable de l’aggravation d’un syndrome hépatorénal. Le foie étant organisé en différents lobules, chaque lobule est drainé par des ramifications des branches de la veine porte qui se jettent ensuite au niveau de la veine centrolobulaire. La veine centrolobulaire reçoit également du sang veineux par les capillaires sinusoïdes qui à la base reçoivent du sang oxygéné grâce à l’artère hépatique.
Lors de la cirrhose, en plus d’une organisation anarchique des hépatocytes, on a une désorganisation artérielle et veineuse ce qui fait qu’un certain nombre des ramifications des branches de la veine porte se retrouvent sclérosés entrainant un manque de drainage du sang veineux venant de la veine porte. Cette dernière, accueillant le même débit sanguin mais ne pouvant pas faire remonter correctement le sang veineux à travers le foie, subira une pression plus importante contre ses parois. On parle alors d’hypertension portale.
Après être passé dans les différents lobules, le sang veineux, s’étant chargé de métabolites relargués par les hépatocytes, se jette dans les veines centrolobulaires qui, en se rejoignant, forment les veines sus hépatiques. La forme modale consiste en la présence de 3 veines sous hépatiques s’abouchant dans la veine cave inférieure (VCI) sous diaphragmatique devenant alors la veine cave inférieure sus-hépatique. Cependant, dans 6 à 10% des cas, il existe une ou plusieurs veines hépatiques droites dites accessoires s’abouchant directement dans la veine cave inférieure et qui drainent les secteurs postérieurs droits hépatiques. Il est également intéressant de remarquer sur la Figure 4 la présence de la veine azygos qui est issue de la réunion des veines œsophagiennes, bronchiques, intercostales et péricardiques, et qui constitue une anastomose entre les veines cave inférieure et supérieure. Cela fait que lorsqu’il y aura un blocage au niveau de la veine cave inférieure, le retour du sang veineux se fera par l’intermédiaire de la veine azygos. Il est enfin primordial de noter qu’il existe, comme on peut le voir en pointillé sur cette Figure, une circulation collatérale entre la veine porte et la veine cave. Celle-ci est médiale et provient de la veine oesocardio-tubérositaire antérieure qui elle-même naît de la veine gastrique gauche. Cette circulation collatérale remonte en haut au niveau des veines œsophagiennes et du plexus veineux périoesophagien qui se jette dans le système azygos ce qui permet le retour du sang vers la veine cave supérieure et enfin au cœur.
B. L’irrigation artérielle hépatique : L’artère hépatique, ou artère hépatique commune, est une grosse artère provenant du tronc cœliaque qui se sépare en 3 embranchements : l’artère gastroduodénale qui irrigue l’estomac, le duodénum et le pancréas, l’artère splénique qui irrigue la rate et l’artère hépatique propre (AHP) qui se sépare elle-même en 2 sous-embranchements qui sont la branche gauche irriguant le foie gauche et la branche droite irriguant le foie droit. D’autres artères hépatiques peuvent être présentes : l’artère hépatique gauche (AHG) qui nait de l’artère gastrique gauche et l’artère hépatique droite (AHD) qui vient de l’artère mésentérique supérieure. Selon les personnes, le nombre d’artères hépatiques peut être différent. Les variations anatomiques observées sont d’ailleurs plus courantes et nombreuses que celles observées dans le système veineux porte. Dans 55 à 65% des cas, la forme modale n’est composée que de l’AHP qui assure à elle seule l’irrigation du foie. Toutefois, il n’est pas rare de trouver des personnes possédant deux ou même les trois artères hépatiques précédemment citées. Ainsi, on retrouve la présence de l’artère hépatique gauche chez 8% des personnes et l’artère hépatique droite dans 11% des cas.
Figure 1C. Variantes anatomiques de la vascularisation hépatique.
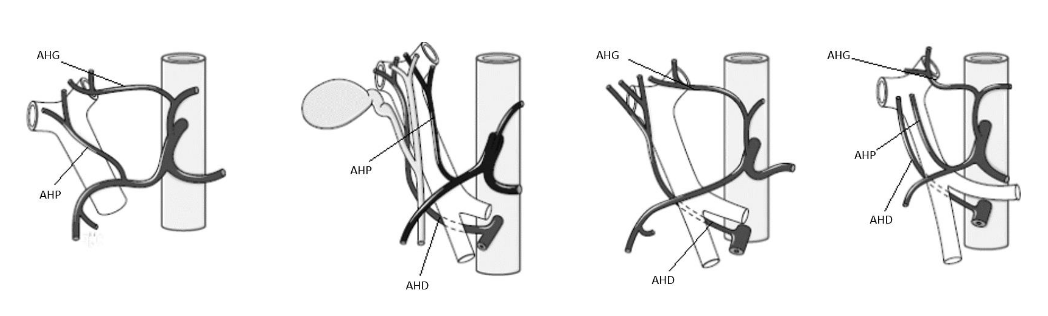 Il existe encore de nombreuses autres variantes comme par exemple l’artère hépatique propre qui naît entièrement de l’artère mésentérique supérieure et non pas du tronc cœliaque comme c’est le cas pour la majorité des personnes. En tout, c’est une dizaine de variantes qui existe concernant la vascularisation du foie par les artères hépatiques et qui ont été classés par Michels en 1955. Ces artères hépatiques permettent non seulement l’apport de dioxygène aux hépatocytes mais également un apport secondaire de xénobiotiques ayant déjà été traités par le foie lors de leur ingestion et leur passage dans la veine porte. D’ailleurs, on peut imaginer que la pharmacocinétique de ces xénobiotiques et notamment le débit, la distribution, la captation par les hépatocytes ou encore la clairance des xénobiotiques peuvent s’avérer différentes selon les variantes anatomiques artérielles et veineuses.
Il existe encore de nombreuses autres variantes comme par exemple l’artère hépatique propre qui naît entièrement de l’artère mésentérique supérieure et non pas du tronc cœliaque comme c’est le cas pour la majorité des personnes. En tout, c’est une dizaine de variantes qui existe concernant la vascularisation du foie par les artères hépatiques et qui ont été classés par Michels en 1955. Ces artères hépatiques permettent non seulement l’apport de dioxygène aux hépatocytes mais également un apport secondaire de xénobiotiques ayant déjà été traités par le foie lors de leur ingestion et leur passage dans la veine porte. D’ailleurs, on peut imaginer que la pharmacocinétique de ces xénobiotiques et notamment le débit, la distribution, la captation par les hépatocytes ou encore la clairance des xénobiotiques peuvent s’avérer différentes selon les variantes anatomiques artérielles et veineuses.
1.3. La segmentation hépatique
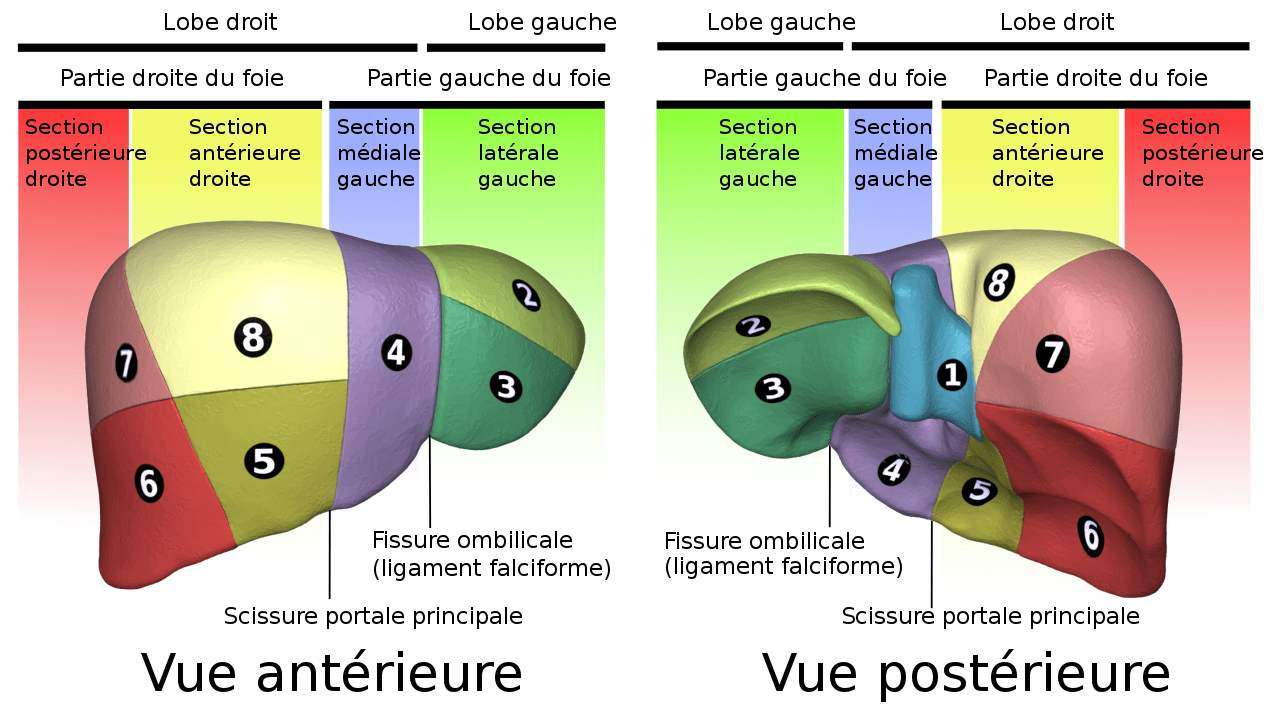
L’étude de ses entités vasculaires est essentielle lorsque l’on veut comprendre l’anatomie fonctionnelle du foie, car la vascularisation du foie permet de distinguer des secteurs qui eux-mêmes sont divisés en segments : c’est la segmentation de Couinaud. Cette segmentation permet la division du foie en 8 segments qui sont répartis, selon leur localisation, au niveau du foie droit ou du foie gauche et qui sont séparés par les embranchements des veines porte et hépatiques. Le foie droit contient les segments V, VI, VII, VIII qui sont vascularisés par la branche droite de l’artère hépatique propre et le foie gauche les segments II, III et IV qui eux sont vascularisés par la branche droite de l’artère hépatique propre. Le segment I, lui, se trouve au niveau postérieur et est partagé entre les deux foies. De par leur localisation, la segmentation de Couinaud se révèle surtout fondamentale en chirurgie bariatrique : l’ablation d’une ou de plusieurs parties du foie suivent souvent les limites anatomiques des différents segments. L’autre intérêt de la segmentation hépatique vient du fait que les segments peuvent répondre différemment à une pathologie hépatique.
Figure 1D. Classification de Couinaud des segments du Foie. © CC BY-SA.
 1.4. Structures hépatiques
1.4. Structures hépatiques
Le foie est constitué principalement d’hépatocytes mais contient également d’autres cellules : (1) des cellules de l’immunité comme des lymphocytes et les cellules de Küppfer qui ne sont d’autres que des macrophages, (2) des cholangiocytes qui sont les cellules épithéliales des conduits biliaires, (3) des cellules endothéliales tapissant les nombreuses voies vasculaires du foie, (4) des cellules de stockage, notamment les cellules de Ito, qui sont des cellules stellaires permettant le stockage de la vitamine A. Ces cellules de Ito, en plus de leur rôle de stockage, sont reconnues comme importantes lors des lésions hépatiques. Les réactions inflammatoires activent et différencient les cellules étoilées en myofibroblastes qui migrent au niveau des lésions et provoquent des changements autant qualitatifs que quantitatifs afin de réparer et restaurer la matrice extracellulaire. C’est l’accumulation excessive de matrice par ces cellules lors d’une inflammation chronique qui est à l’origine de la fibrose hépatique.
Les cellules hépatiques ont la particularité de se regrouper en lobules. Ceux-ci ne sont pas une simple réunion d’hépatocytes, mais c’est tout un réseau ou l’on retrouve un pédicule hépatique (qui vient de la présence de la veine porte avec un canal biliaire et l’artère hépatique), des canalicules acheminant la bile vers le pédicule, des capillaires sinusoïdes permettant les échanges entre les hépatocytes et le sang et une veine centrolobulaire se trouvant, comme son nom l’indique, au centre du lobule et qui permet l’acheminement du sang vers les veines hépatiques. Il est important de souligner le fait que le lobule n’est pas non plus juste un ensemble d’hépatocytes reliés entre eux par des canalicules biliaires ou des capillaires, c’est un ensemble de cellules qui interagissent entre elles afin de donner au foie la capacité de fonctionner. En effet, on remarque la présence des cellules précédemment citées comme les cellules de Küppfer qui permettent la phagocytose de micro-organismes étrangers se trouvant au niveau des capillaires sinusoïdes, les cellules endothéliales se trouvant au niveau des vaisseaux et les cholangiocytes tapissant les canalicules intralobulaires.
1.5. Les voies biliaires
Lors de la digestion lipidique, les hépatocytes rejettent un liquide jaune-verdâtre appelé la bile. Celle-ci est collectée dans les canalicules biliaires qui, anatomiquement, sont séparés des capillaires sinusoïdes d’au moins la moitié de la largeur d’un hépatocyte. Ces canalicules se rejoignent en un canal intermédiaire appelé le canal (ou passage) de Herring qui aboutit au conduit biliaire interlobulaire. La réunion de ces différents conduits interlobulaires permet la formation de deux troncs biliaires au niveau du lobe droit (canal hépatique droit) et du lobe gauche (canal hépatique gauche) du foie qui, en se réunissant, constitue le canal hépatique commun situé au niveau du hile. Une fois arrivée au niveau de ce conduit hépatique commun, la bile continue à descendre le long du canal cholédoque qui provient de la réunion du canal hépatique commun avec le canal cystique. Celui-ci draine la bile qui a été stockée au niveau de la vésicule biliaire à travers des replis qui se dilatent ou se contractent en fonction de la pression que la bile exerce sur la vésicule biliaire. Enfin, la bile descend le canal cholédoque pour rejoindre le canal pancréatique et enfin le duodénum par l’intermédiaire du sphincter d’Oddi. La bile est alors éliminée dans les selles, mais une certaine quantité de cette bile peut être réabsorbée par passage à travers les entérocytes et dans la veine porte ce qui entretient le cycle entérohépatique.
2. Physiologie : les fonctions du foie
2.1. Fonctions nutritionnelles : le métabolisme énergétique
2.1.1. L’homéostasie énergétique (régulation du métabolisme des glucides): Pour mieux appréhender les fluctuations des apports énergétiques, plusieurs organes comme le tissu adipeux, le pancréas et le foie jouent un rôle essentiel dans le maintien de l’homéostasie de l’organisme en participant activement à la régulation du métabolisme glucido-lipidique. Lors de l’ingestion de sucres (prise d’aliments), ceux-ci sont digérés d’une part par les enzymes salivaires mais également par les enzymes gastro-duodénales afin d’être découpés en monosaccharide comme le glucose, le fructose et le galactose. Ceux-ci passant à travers les entérocytes par des transporteurs spécifiques passent au niveau de la veine porte et suivent le réseau veineux pour atterrir au niveau des capillaires sinusoïdes et être absorbés par les hépatocytes.
En post-prandiale, ces monosaccharides peuvent emprunter deux voies métaboliques différentes : la première est la glycogénogénèse qui permet de stocker le glucose sous forme de glycogène rapidement mobilisable en cas de besoins énergétiques. Lorsque cette première voie métabolique est saturée, la seconde voie empruntée par le glucose est la voie de la glycolyse qui permet de générer notamment un pool de protons et d’électrons qui in fine permettra la formation d’énergie sous forme d’ATP. En fait, durant la période post-prandiale, les nutriments tels que le glucose, les acides aminés et les lipides vont passer dans la circulation sanguine et générer une augmentation de la glycémie. Cette augmentation va entrainer au niveau du pancréas une sécrétion d’insuline par les cellules β et une diminution de sécrétion du glucagon par les cellules α. Sous l’action de ces hormones, l’organisme va s’orienter vers un état métabolique particulier, appelé anabolisme, qui permet la transformation et le stockage des glucides et des lipides absorbés lors de la digestion sous la forme respectivement de glycogène et d’acides gras. Le foie durant cette période permet de transformer le glucose, entré dans les hépatocytes via le transporteur GLUT2, en glycogène par le processus de glyconéogenèse.
Le foie permet également de métaboliser le glucose en pyruvate par la glycolyse. Ce pyruvate est ensuite complètement oxydé pour générer de l’énergie sous forme d’adénosine triphosphate (ATP) au cours du cycle de Krebs, suivi de la phosphorylation oxydative (OXPHOS) au sein des mitochondries. Les produits glycolytiques sont également utilisés pour synthétiser des acides gras via la lipogenèse de novo (LDN). Les acides gras sont ensuite estérifiés avec le glycérol-3-phosphate pour former des triglycérides (aussi connus sous le nom de triacylglycérols), ou avec du cholestérol pour former des esters de cholestérols. Les triglycérides et les esters de cholestérols sont stockés dans les gouttelettes lipidiques au sein des hépatocytes ou bien sont sécrétés dans la circulation sous forme de particules de very low density lipoproteine (VLDL). Les acides gras peuvent également être incorporés dans les phospholipides, qui sont les composants essentiels des membranes cellulaires et organites, ainsi que de la couche externe entourant les gouttelettes lipidiques et les VLDL.
En période de jeûne, la diminution de la glycémie va entraîner une inversion du rapport insuline/glucagon, ce qui va orienter l’organisme vers le catabolisme des ressources stockées qui sont donc utilisées pour maintenir une glycémie, à environ 6 mmol/L, et permettre d’alimenter les organes périphériques afin de maintenir leur homéostasie énergétique. À l’état de jeûne, le foie utilise le glycogène stocké pour produire du glucose, lors de la glycogénolyse. Il favorise la synthèse du glucose de novo à partir de lactate, pyruvate, glycérol et d’acides aminées lors de la gluconéogenèse (néoglucogénèse). Le jeûne favorise également la β-oxydation mitochondriale des acides gras (mtFAO pour mitochondrial fatty acid oxidation) qui fournit de l’énergie pour les hépatocytes et permet de générer des corps cétoniques, tel que le β-hydroxybutyrate, l’acétoacétate et l’acétone, lors de la cétogenèse mitochondriale. Les corps cétoniques permettent par la suite de fournir un carburant métabolique pour les tissus extra-hépatiques.
2.1.2. La régulation du métabolisme lipidique par les voies métaboliques énergétiques: Le maintien de l’homéostasie énergétique se fait par différentes voies métaboliques. Ces voies sont finements régulées par de nombreux facteurs de transcription et co-activateurs, qui contrôlent l’expression des enzymes qui catalysent les étapes limitantes de ces voies métaboliques. Ceci permet d’avoir une réponse rapide de l’organisme à chaque signal interne ou externe. Des perturbations de ces voies métaboliques peuvent conduire à un métabolisme énergétique aberrant et favoriser l’apparition de maladies telles que la résistance à l’insuline, le diabète et des lésions hépatiques. Les principales voies métaboliques sont :
A. Les voies métaboliques mitochondriales : La mitochondrie est un organite indispensable à la vie, notamment de par sa capacité à produire l’énergie nécessaire pour l’ensemble des cellules du corps sous forme d’ATP. Le nombre de mitochondries présentes dans les cellules est variable selon les tissus, et souvent corrélé à son activité métabolique : ainsi nous pouvons trouver de nombreuses mitochondries dans les muscles, le cœur, le cerveau et le foie. La mitochondrie interagit avec de nombreuses voies métaboliques qui peuvent pour certaines avoir lieu au sein même de celle-ci. En effet, la mitochondrie joue un rôle dans l’étape finale de dégradation des glucides, protéines et lipides, avec la glycolyse qui aboutit à la formation de pyruvate qui est ensuite converti en acétyl-CoA au niveau mitochondrial. La β-oxydation des acides gras, qui se déroule majoritairement au sein des mitochondries, conduit également à la formation d’acétyl-CoA. Cet acétyl-CoA est ensuite oxydé dans la mitochondrie par le cycle de Krebs et permet la formation de coenzymes réduits NADH et FADH2 qui sont utilisés par la chaîne respiratoire mitochondriale pour fournir de l’ATP (Figure 2).
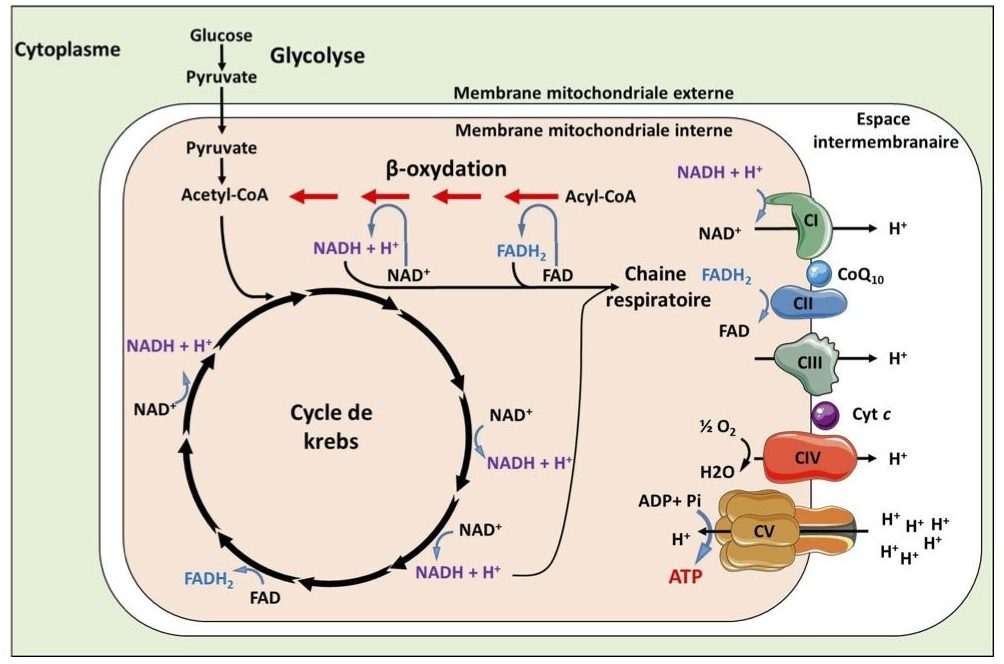 Figure 2. Schéma simplifié du métabolisme mitochondriale. La dégradation du glucose par la glycolyse et le cycle de Krebs génère une réduction du NAD+ (nicotinamide adénine dinucléotide) et FAD (flavine adénine dinucléotide) pour former du NADH et du FADH2. La β-oxydation mitochondriale des esters d’acyl-CoA est réalisée par quatre réactions enzymatiques qui génèrent également du NADH et FADH2, ainsi que de l’acétyl-CoA qui peut à son tour rentrer dans le cycle de Krebs pour produire les cofacteurs réduits. Les électrons dérivés du NADH et FADH2 sont utilisés par les cinq complexes de la chaine respiratoire pour générer de l’ATP. Le complexe I (NADH déshydrogénase), complexe III (coenzyme Q-cytochrome c réductase) et le complexe IV (cytochrome c oxydase) pompent les protons (H+) hors de la matrice mitochondriale pour générer un potentiel de membrane. Ce potentiel est utilisé par le complexe V (ATP synthase) pour former de l’ATP. © Adapté de (Nsiah-Sefaa et McKenzie, 2016) Pi : phosphate inorganique ; Complexe II (succinate déshydrogénase).
Figure 2. Schéma simplifié du métabolisme mitochondriale. La dégradation du glucose par la glycolyse et le cycle de Krebs génère une réduction du NAD+ (nicotinamide adénine dinucléotide) et FAD (flavine adénine dinucléotide) pour former du NADH et du FADH2. La β-oxydation mitochondriale des esters d’acyl-CoA est réalisée par quatre réactions enzymatiques qui génèrent également du NADH et FADH2, ainsi que de l’acétyl-CoA qui peut à son tour rentrer dans le cycle de Krebs pour produire les cofacteurs réduits. Les électrons dérivés du NADH et FADH2 sont utilisés par les cinq complexes de la chaine respiratoire pour générer de l’ATP. Le complexe I (NADH déshydrogénase), complexe III (coenzyme Q-cytochrome c réductase) et le complexe IV (cytochrome c oxydase) pompent les protons (H+) hors de la matrice mitochondriale pour générer un potentiel de membrane. Ce potentiel est utilisé par le complexe V (ATP synthase) pour former de l’ATP. © Adapté de (Nsiah-Sefaa et McKenzie, 2016) Pi : phosphate inorganique ; Complexe II (succinate déshydrogénase).
A.1. La β-oxydation mitochondriale : L’oxydation des acides gras peut se dérouler dans trois compartiments cellulaires, avec la β- oxydation qui peut se dérouler dans le peroxysome et la mitochondrie et la ω-oxydation dans le réticulum endoplasmique (RE). La β-oxydation mitochondriale des acides gras (mtFAO, pour mitochondrial fatty acid oxidation) permet de faciliter la dégradation des acides gras (AG), préalablement activés en acétyl-coenzyme A (acétyl-CoA), et ainsi permettre d’obtenir de l’énergie rapidement et efficacement. Par exemple, l’oxydation d’une molécule de palmitate (un acide gras à chaîne longue de 16 carbones) produit jusqu’à 129 équivalents d’ATP.
Cette voie métabolique est présentée Figure 3. Tout d’abord, les acides gras peuvent entrer dans les cellules hépatiques depuis la circulation, par diffusion pour les AG à chaînes courtes (AGCC, < C6) et moyennes (AGCM, C8-C12) ou via des transporteurs pour les AG à chaînes longues (AGCL, C14- C20) et très longues (AGCTL, >22). Les principaux transporteurs sont la fatty acid translocase/cluster of differentiation 36 (FAT/CD36) et les membres de la famille des fatty acid transport protein (FATPs) avec majoritairement FATP2 et FATP5 pour le foie. Il est à noter que les AGCTL ne semblent pas être métabolisés dans la mitochondrie et sont plutôt oxydés dans les peroxysomes par un processus similaire. La première étape de la mtFAO est l’entrée des AG dans la mitochondrie. Les AGCC et AGCM peuvent librement traverser la membrane mitochondriale et être activés par l’acyl-CoA-synthétase (ACS) en acyl-CoA dans la matrice mitochondriale. Tandis que les AGCL requièrent un système de navette spécifique qui se déroule en 4 étapes.
Premièrement, l’activation des AGCL en acyl-CoA se fait par l’ACS, présente dans le cytosol. Les acyl-CoA sont ensuite convertis en acyl-carnitine par la carnitine palmitoyltransférase 1 (CPT1), localisé du coté cytosolique de la membrane externe de la mitochondrie. Les acyl-carnitine peuvent à présent être transloqués à travers la membrane interne de la mitochondrie via la carnitine-acylcarnitine translocase (CACT). Enfin, la carnitine palmitoyltransférase 2 (CPT2) localisée sur la face interne de la membrane interne de la mitochondrie, permet l’échange de la carnitine par la CoA pour permettre de retrouver l’acyl-CoA. Les acyl-CoA présents dans la matrice de la mitochondrie peuvent subir les cycles de β-oxydation (aussi appelé hélice de Lynen), qui s’effectuent en 4 réactions successives. La première réaction du cycle de β-oxydation est réalisée par des acyl-CoA déshydrogénases spécifiques selon la longueur de chaîne des acyl-CoA et va aboutir à la formation de trans-Δ2-énoyl-CoA et du cofacteur FADH2 (utilisé par le complexe II de la chaîne respiratoire). Il y a ensuite une hydratation, par l’énoyl-CoA hydratase, qui va générer la L-3-hydroacyl-CoA. La troisième réaction est réalisée par la 3-hydroacyl-CoA déshydrogénase pour permettre la formation du β-cétoacyl-CoA et du cofacteur NADH (utilisé par le complexe I de la chaîne respiratoire). Enfin, le β- cétoacyl-CoA est utilisé par la β-cétothiolase pour former de l’acétyl-CoA et de l’acyl-CoA raccourcie de 2 carbones, qui pourra rentrer dans un nouveau cycle de β-oxydation.
Dans le foie, une part significative des acétyl-CoA formés lors de la β-oxydation est utilisée par la cétogenèse pour générer des corps cétoniques, comme l’acétoacétate, l’acétone et le β- hydroxybutyrate, surtout en phase de jeûne. Les corps cétoniques sont par la suite relargués dans la circulation sanguine pour être utilisés par les tissus extra-hépatiques afin de leur fournir de l’énergie.
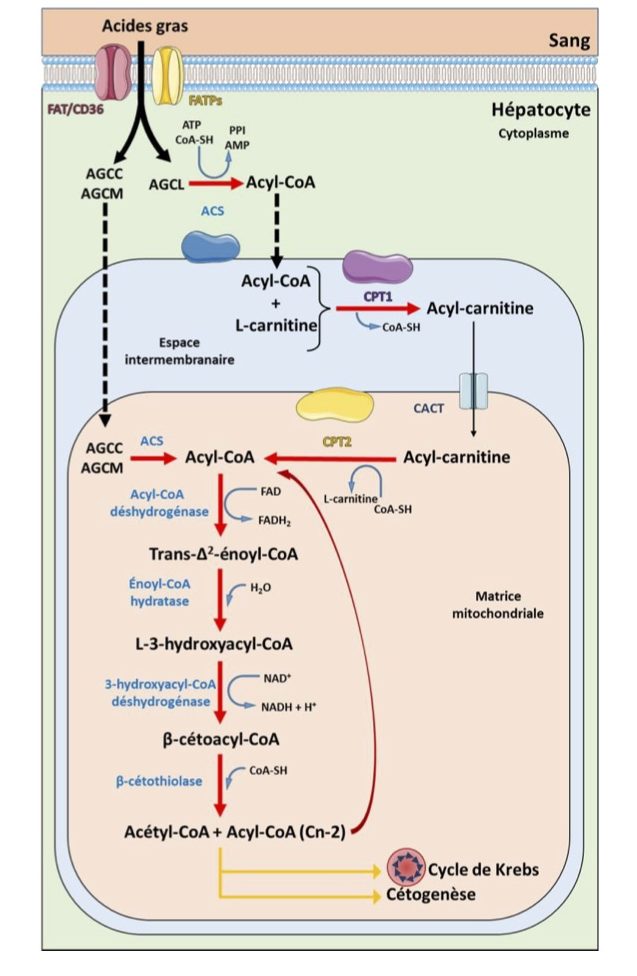 Figure 3. Schéma de la β-oxydation mitochondriale des acides gras. Le transport des acides gras à travers la membrane plasmique est réalisé par différents transporteurs FAT/CD36 (fatty acid translocase/cluster of differenciation 36) et FATPs (fatty acid transport protein). Tandis que les acides gras à chaînes courtes (AGCC) et moyennes (AGCM) diffusent à travers les membranes mitochondriales, les acides gras à chaîne longue (AGCL) sont activés en acyl-CoA par l’acyl-CoA synthétase (ACS). La CPT1 (carnitine palmitoyltransférase 1) convertit l’acyl-CoA en acylcarnitine, qui peut ensuite être transporté à travers la membrane mitochondriale par la carnitine-acylcarnitine translocase (CACT). CPT2 convertit l’acyl-carnitine de nouveau en acyl-CoA. Les acides gras à chaînes courtes et moyennes traversent les membranes mitochondriales par diffusion, et sont activés dans la matrice mitochondriale. Les acyl-CoAs sont ensuite métabolisées en 4 réactions successives par différentes enzymes selon la taille des acyl-CoA. Durant ces étapes, des cofacteurs sont générés ainsi qu’un acétyl-CoA et un acyl-CoA raccourcie de deux carbones. L’acétyl-CoA peut ensuite entrer dans le cycle de Krebs ou dans la voie de la cétogenèse. Adapté de (Fromenty, 2019; Houten et al., 2016; Rui, 2014; Schönfeld et Wojtczak, 2016; Wanders et al., 2010).
Figure 3. Schéma de la β-oxydation mitochondriale des acides gras. Le transport des acides gras à travers la membrane plasmique est réalisé par différents transporteurs FAT/CD36 (fatty acid translocase/cluster of differenciation 36) et FATPs (fatty acid transport protein). Tandis que les acides gras à chaînes courtes (AGCC) et moyennes (AGCM) diffusent à travers les membranes mitochondriales, les acides gras à chaîne longue (AGCL) sont activés en acyl-CoA par l’acyl-CoA synthétase (ACS). La CPT1 (carnitine palmitoyltransférase 1) convertit l’acyl-CoA en acylcarnitine, qui peut ensuite être transporté à travers la membrane mitochondriale par la carnitine-acylcarnitine translocase (CACT). CPT2 convertit l’acyl-carnitine de nouveau en acyl-CoA. Les acides gras à chaînes courtes et moyennes traversent les membranes mitochondriales par diffusion, et sont activés dans la matrice mitochondriale. Les acyl-CoAs sont ensuite métabolisées en 4 réactions successives par différentes enzymes selon la taille des acyl-CoA. Durant ces étapes, des cofacteurs sont générés ainsi qu’un acétyl-CoA et un acyl-CoA raccourcie de deux carbones. L’acétyl-CoA peut ensuite entrer dans le cycle de Krebs ou dans la voie de la cétogenèse. Adapté de (Fromenty, 2019; Houten et al., 2016; Rui, 2014; Schönfeld et Wojtczak, 2016; Wanders et al., 2010).
La régulation de la mtFAO est complexe et implique de nombreux facteurs dont les principaux sont présentés figure 4. Les enzymes impliquées dans la mtFAO et la cétogenèse sont positivement régulées au niveau transcriptionnel par peroxisome proliferator-activated receptor (PPAR) α, un récepteur nucléaire et facteur de transcription qui peut être activé par différent AG naturels ou médicaments tels que les fibrates. D’autres facteurs de transcription peuvent également réguler positivement la mtFAO, on trouve PPAR β/δ, forkhead box A2 (FoxA2), cAMP-response element-binding protein (CREB) et hepatocyte nuclear factor 4 α. De plus PPARγ coactivator-1α (PGC-1α) joue également un rôle d’activateur clé dans la régulation transcriptionnelle des enzymes de la mtFAO. Il existe également des régulations post-transcriptionnelles avec par exemple l’inhibition de la CPT1 par le malonyl-CoA, un intermédiaire de la lipogenèse de novo.
![]() Figure 4. Schéma des protéines et facteurs de transcription régulant la beta-oxydation. Abréviations : AG : acides gras ; PPAR : peroxisome proliferator-activated receptor ; ERRα : Estrogen- related receptor alpha ; IL-6 : interleukine-6 ; CREB : cAMP-response element-binding protein ; PGC- 1α : PPARγ coactivator-1α ; SIRT : sirtuine 1 ; RXR : retinoid X receptor ; HNF4α : hepatocyte nuclear factor 4 α ; FoxA2 : forkhead box A2 ; NF-κB : nuclear factor-kappa B ; JNK : c-Jun N-terminal kinases ; AMP : Adénosine monophosphate ; AMPK : AMP-activated protein kinase ; ACC : acétyl-coenzyme A carboxylase ; CD36 : cluster of differentiation 36 ; ACS : Acetyl-coenzyme A synthetase ; ACOX : Peroxisomal acyl-coenzyme A oxidase ; CPT1 : Carnitine palmitoyltransferase I ; FABP : fatty-acid- binding proteins ; FATP : fatty acid transport protein ; 3-KAT : acétyl-CoA acétyltransferase ; MCD : malonyl-CoA decarboxylase ; VLCAD, LCAD, MCAD, SCAD : very-long-, long-, medium-, short-chain acyl- CoAdehydrogenase; GPAT:glycerol3-phosphateacyltransferase; FGF21: fibroblastgrowthfactor 21. © Adaptation de (Drosatos et Schulze, 2013).
Figure 4. Schéma des protéines et facteurs de transcription régulant la beta-oxydation. Abréviations : AG : acides gras ; PPAR : peroxisome proliferator-activated receptor ; ERRα : Estrogen- related receptor alpha ; IL-6 : interleukine-6 ; CREB : cAMP-response element-binding protein ; PGC- 1α : PPARγ coactivator-1α ; SIRT : sirtuine 1 ; RXR : retinoid X receptor ; HNF4α : hepatocyte nuclear factor 4 α ; FoxA2 : forkhead box A2 ; NF-κB : nuclear factor-kappa B ; JNK : c-Jun N-terminal kinases ; AMP : Adénosine monophosphate ; AMPK : AMP-activated protein kinase ; ACC : acétyl-coenzyme A carboxylase ; CD36 : cluster of differentiation 36 ; ACS : Acetyl-coenzyme A synthetase ; ACOX : Peroxisomal acyl-coenzyme A oxidase ; CPT1 : Carnitine palmitoyltransferase I ; FABP : fatty-acid- binding proteins ; FATP : fatty acid transport protein ; 3-KAT : acétyl-CoA acétyltransferase ; MCD : malonyl-CoA decarboxylase ; VLCAD, LCAD, MCAD, SCAD : very-long-, long-, medium-, short-chain acyl- CoAdehydrogenase; GPAT:glycerol3-phosphateacyltransferase; FGF21: fibroblastgrowthfactor 21. © Adaptation de (Drosatos et Schulze, 2013).
A.2. La chaîne respiratoire mitochondriale : La chaîne respiratoire mitochondriale est associée à différentes voies métaboliques pour aboutir à la production d’énergie sous forme d’ATP, avec par exemple un lien très étroit entre la chaîne respiratoire et la mtFAO. En effet, la chaîne respiratoire mitochondriale a pour rôle de réoxyder les coenzymes réduits NADH et FADH2, issus de la mtFAO, du cycle de Krebs et de la glycolyse permettant ainsi de fournir de l’énergie sous forme d’ATP à la cellule. Ce phénomène constitue le processus de phosphorylation oxydative (OXPHOS). Les électrons, contenus dans ces coenzymes réduits, fournissent l’énergie libre nécessaire pour pomper des protons de la matrice de la mitochondrie vers l’espace intermembranaire. Le complexe I (NADH déshydrogénase) oxyde le NADH en NAD+ + H+ et transfère ainsi deux électrons à l’ubiquinone (ou coenzyme Q10) en la réduisant en ubiquinol.
Simultanément, le complexe II (succinate déshydrogénase), l’enzyme qui relie le cycle de Krebs à l’OXPHOS, oxyde le FADH2 en FAD + 2H+ et réduit l’ubiquinone, tout en oxydant le succinate en fumarate tous deux intermédiaires du cycle de Krebs. À partir de l’ubiquinol, les électrons sont transportés vers le complexe III (coenzyme Q- cytochrome c réductase), ce qui facilite le transfert d’électrons vers le cytochrome c, qui à son tour réduit le complexe IV (cytochrome c oxydase). Enfin, les électrons sont transférés à l’oxygène moléculaire (O2) permettant la formation d’eau (H2O). Lorsque le rapport ADP/ATP tend à augmenter dans la cellule, les protons sont réinternalisés dans la matrice mitochondriale à travers la partie F0 de l’ATP synthase (complexe V) et l’énergie libérée est utilisée par la partie F1 de ce même complexe pour synthétiser de l’ATP (Figure 5). Les cinq complexes multimériques qui composent la phosphorylation oxydative mitochondriale sont intégrés dans la membrane interne de la mitochondrie. Ces complexes sont constitués de nombreuses sous-unités (environ 90 protéines), qui peuvent être codées par l’ADN mitochondriale ou par l’ADN nucléaire. 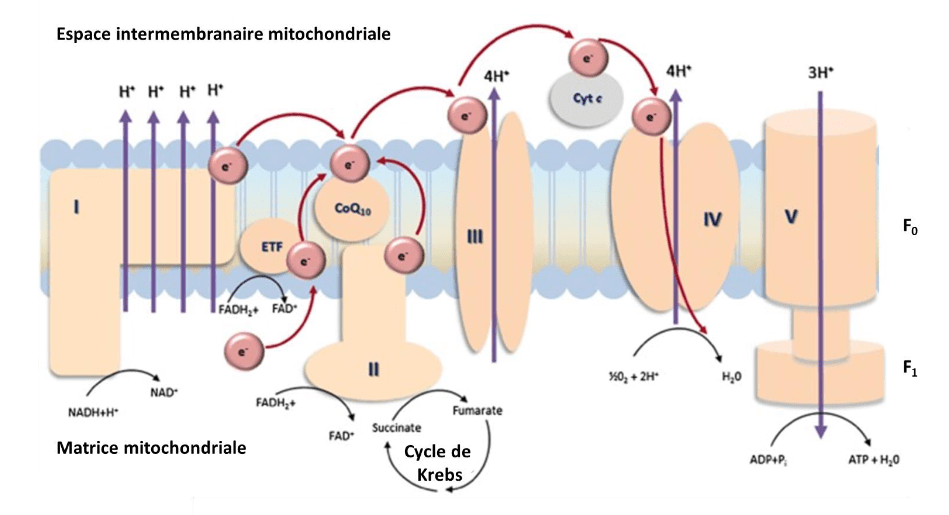 Figure 5 : Schéma de la phosphorylation oxydative mitochondriale. Le complexe I (NADH déshydrogénase) oxyde le NADH en NAD+ et H+ et transmet deux électrons à l’ubiquinone (CoQ10) le réduisant en ubiquinol. Simultanément, le complexe II (succinate déshydrogénase), l’enzyme qui relie le cycle de Krebs à l’OXPHOS, oxyde le FADH en FAD+ et H+ et réduit l’ubiquinone, tout en oxydant le succinate en fumarate, deux intermédiaires du cycle de Krebs. De l’ubiquinol, les électrons sont transportés vers le complexe III (coenzyme Q-cytochrome c réductase), ce qui facilite le transfert des électrons vers le cytochrome c, qui à son tour réduit le complexe IV (cytochrome c oxydase). Enfin les électrons sont donnés à l’oxygène moléculaire et de l’eau se forme. Lorsque le rapport ADP/ATP tend à augmenter dans la cellule, les protons sont réinternalisés dans la matrice mitochondriale à travers la partie F0 du complexe V (ATP synthase) et l’énergie libérée est utilisée par la partie F1 de ce même complexe pour synthétiser de l’ATP. © Adapté de (Kanabus et al., 2014).
Figure 5 : Schéma de la phosphorylation oxydative mitochondriale. Le complexe I (NADH déshydrogénase) oxyde le NADH en NAD+ et H+ et transmet deux électrons à l’ubiquinone (CoQ10) le réduisant en ubiquinol. Simultanément, le complexe II (succinate déshydrogénase), l’enzyme qui relie le cycle de Krebs à l’OXPHOS, oxyde le FADH en FAD+ et H+ et réduit l’ubiquinone, tout en oxydant le succinate en fumarate, deux intermédiaires du cycle de Krebs. De l’ubiquinol, les électrons sont transportés vers le complexe III (coenzyme Q-cytochrome c réductase), ce qui facilite le transfert des électrons vers le cytochrome c, qui à son tour réduit le complexe IV (cytochrome c oxydase). Enfin les électrons sont donnés à l’oxygène moléculaire et de l’eau se forme. Lorsque le rapport ADP/ATP tend à augmenter dans la cellule, les protons sont réinternalisés dans la matrice mitochondriale à travers la partie F0 du complexe V (ATP synthase) et l’énergie libérée est utilisée par la partie F1 de ce même complexe pour synthétiser de l’ATP. © Adapté de (Kanabus et al., 2014).
B. La lipogenèse de novo : La lipogenèse de novo (LDN) est la voie métabolique de la synthèse des acides gras. Elle se déroule principalement dans le foie et préférentiellement après un repas riche en glucides. Ces acides gras peuvent ensuite être incorporés dans des triglycérides et permettre un stockage d’énergie. Cette voie métabolique, présentée figure 6, implique plusieurs autres voies métaboliques telles que la glycolyse qui, à partir du glucose, aboutit à la formation de pyruvate. Celui-ci est ensuite utilisé par la pyruvate déshydrogénase (PDH), présente dans la mitochondrie, pour former de l’acétyl-CoA. L’acétyl-CoA entre ensuite dans le cycle de Krebs pour former du citrate, lors de la première étape du cycle. Le citrate est ensuite exporté en dehors de la mitochondrie où il est métabolisé, dans le cytoplasme, par l’ATP-citrate lyase (ACLY) pour former de l’acétyl-CoA cytoplasmique, ainsi que de l’oxaloacétate. Ce dernier peut être réduit en malate, puis converti en pyruvate avec un relargage de NADPH. L’acétyl-CoA est carboxylé par l’acétyl-CoA carboxylase (ACC) pour former du malonyl-CoA. Le malonyl-CoA et la NADPH sont utilisés comme précurseurs par l’acide gras synthase (FAS, anglais fatty acid synthase) pour la synthèse de palmitate (C16). Il peut s’ensuivre une élongation des acides gras, réalisée par l’acyl-CoA élongase (Elovl, anglais elongation of very long chain fatty acids), pour générer des AGCL (>16C). Les AGCL peuvent être pris en charge par des protéines de la famille des stéroyl-CoA désaturases (SCDs) pour être transformés en acides gras insaturés (AGI). L’ensemble de ces acides gras nouvellement formés peut ensuite être stocké sous forme de triglycérides et incorporé dans des gouttelettes lipidique pour constituer une réserve d’énergie ou être exporté en dehors de la cellule sous forme de VLDL pour fournir de l’énergie aux organes extra-hépatiques.
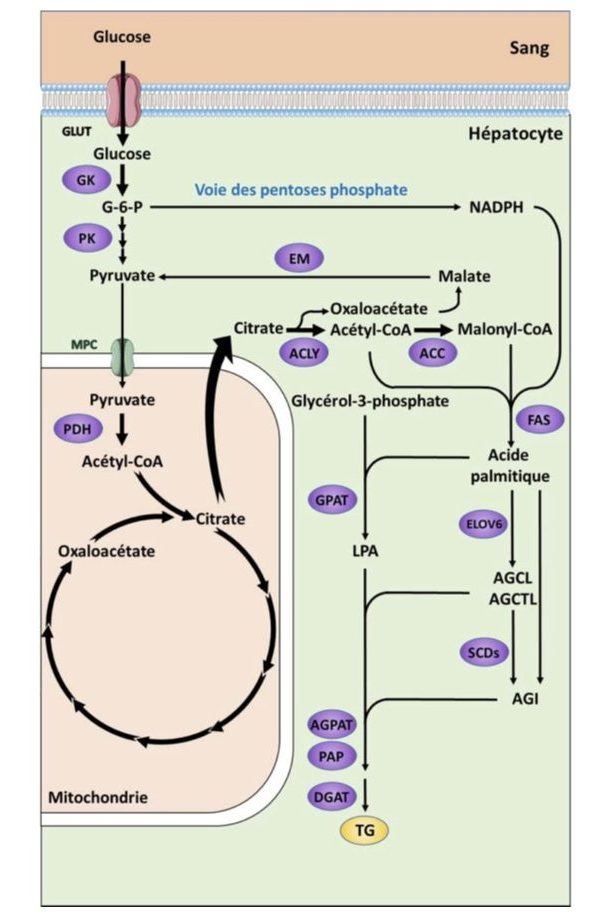 Figure 6. Schéma de la lipogenèse de novo et de la synthèse des triglycérides. Le glucose absorbé par la cellule est converti en pyruvate par glycolyse. Le pyruvate est ensuite converti en acétyl-CoA et entre dans le cycle de Krebs via le transporteur mitochondrial pyruvate carrier (MPC). Une fois dans la mitochondrie le pyruvate est converti de nouveau en acétyl-CoA par la pyruvate deshydrogenase (PDH). Un excès d’acétyl-CoA converti en citrate peut sortir de la mitochondrie et devenir le substrat des enzymes lipogéniques. Les enzymes impliquées dans la synthèse des acides gras et des triglycérides (TG) comprennent : (A) les enzymes glycolytiques telles que la glucokinase (GK) et la pyruvate kinase hépatique (PK) qui fournissent la source de carbone pour les acides gras et la synthèse des triglycérides.
- les enzymes pour la voie de synthèse des acides gras, telle que ATP-citrate lyase (ACLY), l’acétyl-CoA carboxylase (ACC), fatty acid synthase (FAS), elongase of long chain fatty acids family 6 (ELOV6) et le stéroyl-CoA désaturase (SCD) pour former des acides gras insaturés (AGI), (B) enzyme pour la production de NADPH utilisée dans la synthèse des acides gras, comprenant la branche oxydative de la voie des pentoses phosphate et l’enzyme malique (EM).
- les enzymes impliquées dans l’estérification pour la production de TG, telles que la glycerol-3- phosphate acyltransférase (GPAT), 1-acylglycerol-3-phosphate acyltransférase (AGPAT), phosphatidate phosphatase (PAP) et diacylglycerol acyltransferase (DGAT). © Adapté de (Rui, 2014; Yuhui Wang et al., 2015).
Figure 6. Schéma de la lipogenèse de novo et de la synthèse des triglycérides. Le glucose absorbé par la cellule est converti en pyruvate par glycolyse. Le pyruvate est ensuite converti en acétyl-CoA et entre dans le cycle de Krebs via le transporteur mitochondrial pyruvate carrier (MPC). Une fois dans la mitochondrie le pyruvate est converti de nouveau en acétyl-CoA par la pyruvate deshydrogenase (PDH). Un excès d’acétyl-CoA converti en citrate peut sortir de la mitochondrie et devenir le substrat des enzymes lipogéniques. Les enzymes impliquées dans la synthèse des acides gras et des triglycérides (TG) comprennent : (A) les enzymes glycolytiques telles que la glucokinase (GK) et la pyruvate kinase hépatique (PK) qui fournissent la source de carbone pour les acides gras et la synthèse des triglycérides.
- les enzymes pour la voie de synthèse des acides gras, telle que ATP-citrate lyase (ACLY), l’acétyl-CoA carboxylase (ACC), fatty acid synthase (FAS), elongase of long chain fatty acids family 6 (ELOV6) et le stéroyl-CoA désaturase (SCD) pour former des acides gras insaturés (AGI), (B) enzyme pour la production de NADPH utilisée dans la synthèse des acides gras, comprenant la branche oxydative de la voie des pentoses phosphate et l’enzyme malique (EM).
- les enzymes impliquées dans l’estérification pour la production de TG, telles que la glycerol-3- phosphate acyltransférase (GPAT), 1-acylglycerol-3-phosphate acyltransférase (AGPAT), phosphatidate phosphatase (PAP) et diacylglycerol acyltransferase (DGAT). © Adapté de (Rui, 2014; Yuhui Wang et al., 2015).
La LDN est contrôlée principalement par la régulation de la transcription des gènes de la glycolyse et de la lipogenèse, mais aussi par certains gènes et facteurs de transcription impliqués dans la régulation de l’absorption, du trafic et/ou du stockage des lipides (figure 7). Le facteur de transcription carbonhydrate response element-binding protein (ChREBP) se fixe et active le promoteur de la pyruvate kinase (PK) dans les hépatocytes, une enzyme clé de la glycolyse. ChREBP induit également l’expression de protéines de la LDN comme ACLY, ACC, FAS, SCD1, Elovls et EM (enzyme malique). ChREBP est régulé négativement par le glucagon, qui va stimuler sa phosphorylation sur sa Serine 196 et induire son inactivation par export nucléaire.
À l’opposé, ChREBP est régulé positivement par le glucose qui va induire une acétylation en lysine 672 pour augmenter l’activité de ChREBP. De plus, des intermédiaires de la glycolyse, tel que le glucose-6-phosphate (G6P) ou fructose-2,6-phosphate (F2,6P), peuvent également stimuler l’activité de ChREBP. Les facteurs de transcription de la famille des sterol regulatory element-binding proteins (SREBPs), en particulier SREBP-1c, sont principalement stimulés par l’insuline pour réguler des enzymes clés du métabolisme des lipides. En effet, SREBP-1c peut être activé par clivage dans l’appareil de Golgi et par la suite induire l’expression des gènes de la LDN, comme FAS. PGC-1β joue également un rôle de co-activateur pour SREBP et stimule la LDN. Les récepteurs nucléaires liver X receptor (LXR) et farnesoid X receptor (FXR) peuvent respectivement réguler positivement ou négativement la LDN via le contrôle de l’expression de SREBP1 et ChREBP. Par ailleurs, PPARγ, un facteur de transcription important dans le métabolisme peut stimuler l’expression de nombreux gènes contrôlant l’entrée des acides gras, leur trafic dans la cellule et la biosynthèse des triglycérides.
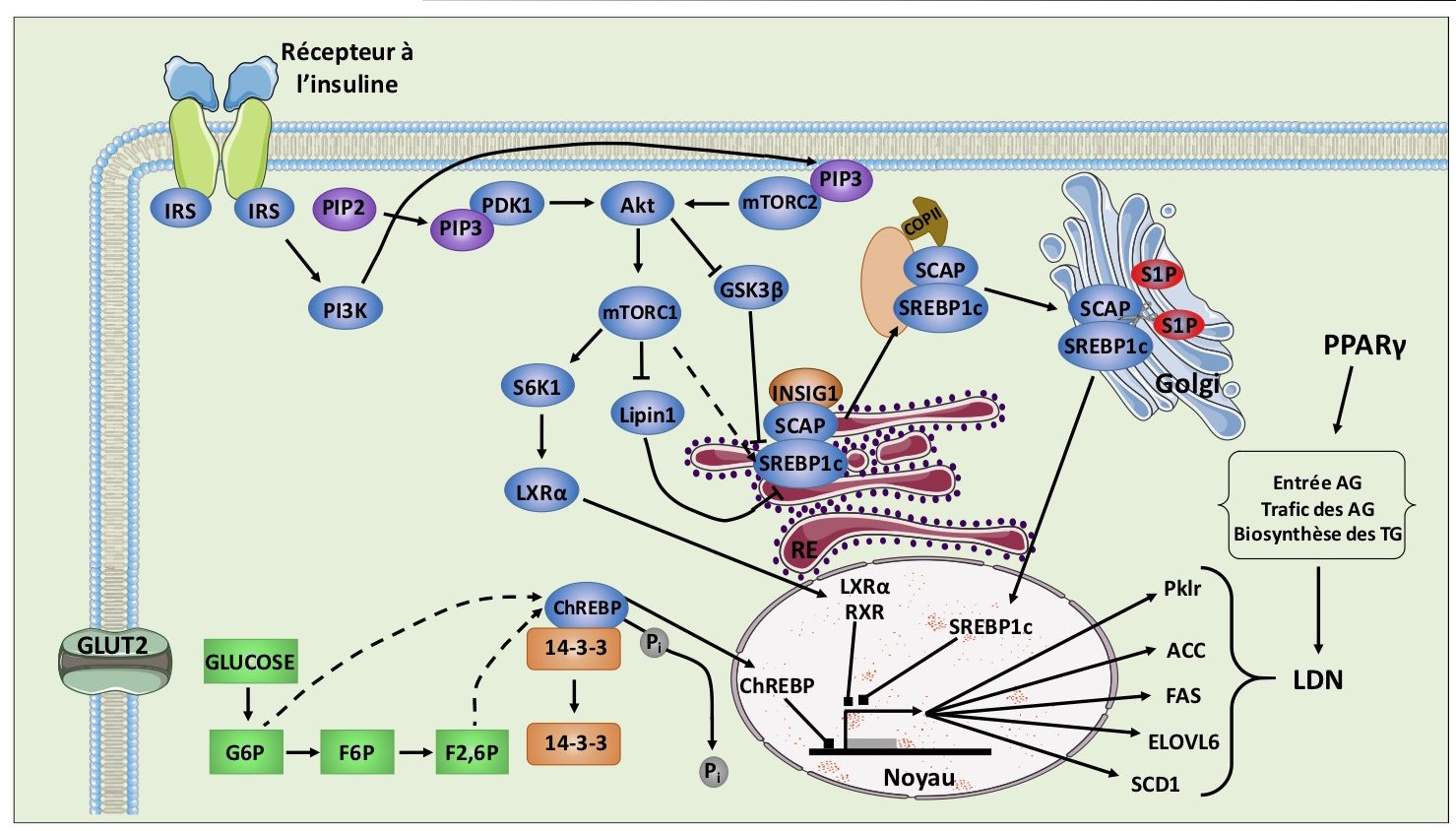 Figure 7 : Schéma de la régulation de la lipogenèse de novo. L’activation du récepteur à l’insuline par l’insuline entraine la phosphorylation de insulin receptor substrate 1 (IRS1), qui active ensuite la phosphoinositide 3- kinase (PI3K), conduisant à la phosphorylation du phosphatidylinositol (4,5)-biphosphate (PIP2) pour former du phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 active à la fois phosphoinositide-dependent kinase 1 (PDK1) et mammalian target of rapamycin complex 2 (mTORC2), qui vont ensuite phosphoryler la protein kinase B (PKB). La PKB peut activer mTORC1 et conduire à l’activation de ribosomal protein S6 kinase 1 (S6K1) et conduire à la relocalisation nucléaire de liver X receptor α (LXRα), et son hétérodimerisation avec RXR pour permettre la transcription de gènes de la lipogenèse de novo (LDN) dont sterol regulatory element binding protein 1c (SREBP1c). La maturation de SREBP1c est favorisée par l’activité de mTORC1 qui permet l’inhibition de lipin 1 mais également par l’inhibition de glycogen synthase kinase 3β (GSK3β) par PKB. Le SREBP1c naissant est associé dans le réticulum endoplasmique (RE) à SREBP cleavage activated protein (SCAP) et insulin-induced gene 1 (INSIG1). INSIG1 inhibe la navette médiée par SCAP de SREBP1c vers le Golgi via des vésicules recouvertes de coat protein II (COPII). Lorsque SREBP1c et SCAP sont phosphorylés ou que des stérols se lient à SCAP, un changement de conformation s’opère dissociant INSIG1 et permettant l’interaction avec les protéines COPII. SREBP1c est ensuite clivé par site 1-protease (S1P) et site 2-protease (S2P) dans le Golgi conduisant à la dissociation de SREBP1c de SCAP et à l’élimination du domaine transmembranaire pour permettre à la forme mature de SREBP1c de se localiser dans le noyau. Une fois dans le noyau, SREBP1c induit la transcription des gènes de la LDN, tels que fatty acid synthase (FAS), stearoyl-CoA desaturase 1 (SCD1), elongation of long-chain fatty acids family member 6 (ELOVL6), and acetyl CoA carboxylase (ACC). Par ailleurs, le glucose peut entrer dans les hépatocytes via glucose transporter 2 (GLUT2) dans les hépatocytes et entrer dans la voie de la glycolyse. Le glucose est phosphorylé en glucose 6-phosphate (G6P) suivi par son isomerisation en fructose 6-phosphate (F6P) et par la suite phosphorylé en fructose-2,6-bisphosphate (F2,6P). G6P et F2,6P peuvent conduire à la dephosphorylation du carbohydrate response element binding protein (ChREBP) et la dissociation de la protéine 14-3-3. Ceci permet la localisation nucléaire du ChREBP et la transcription de ses gènes cibles de la LDN dont FAS, SCD1, ELVL6, ACC et le gène de la pyruvate kinase L/R (Pklr). PPARγ permet l’activation de différents gènes impliqués dans le transport des acides gras (AG) et dans la biosynthèse des triglycérides (TG) favorisant ainsi la LDN et la mise en place d’une stéatose hépatique. © Adapté de (Sanders et Griffin, 2016).
Figure 7 : Schéma de la régulation de la lipogenèse de novo. L’activation du récepteur à l’insuline par l’insuline entraine la phosphorylation de insulin receptor substrate 1 (IRS1), qui active ensuite la phosphoinositide 3- kinase (PI3K), conduisant à la phosphorylation du phosphatidylinositol (4,5)-biphosphate (PIP2) pour former du phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 active à la fois phosphoinositide-dependent kinase 1 (PDK1) et mammalian target of rapamycin complex 2 (mTORC2), qui vont ensuite phosphoryler la protein kinase B (PKB). La PKB peut activer mTORC1 et conduire à l’activation de ribosomal protein S6 kinase 1 (S6K1) et conduire à la relocalisation nucléaire de liver X receptor α (LXRα), et son hétérodimerisation avec RXR pour permettre la transcription de gènes de la lipogenèse de novo (LDN) dont sterol regulatory element binding protein 1c (SREBP1c). La maturation de SREBP1c est favorisée par l’activité de mTORC1 qui permet l’inhibition de lipin 1 mais également par l’inhibition de glycogen synthase kinase 3β (GSK3β) par PKB. Le SREBP1c naissant est associé dans le réticulum endoplasmique (RE) à SREBP cleavage activated protein (SCAP) et insulin-induced gene 1 (INSIG1). INSIG1 inhibe la navette médiée par SCAP de SREBP1c vers le Golgi via des vésicules recouvertes de coat protein II (COPII). Lorsque SREBP1c et SCAP sont phosphorylés ou que des stérols se lient à SCAP, un changement de conformation s’opère dissociant INSIG1 et permettant l’interaction avec les protéines COPII. SREBP1c est ensuite clivé par site 1-protease (S1P) et site 2-protease (S2P) dans le Golgi conduisant à la dissociation de SREBP1c de SCAP et à l’élimination du domaine transmembranaire pour permettre à la forme mature de SREBP1c de se localiser dans le noyau. Une fois dans le noyau, SREBP1c induit la transcription des gènes de la LDN, tels que fatty acid synthase (FAS), stearoyl-CoA desaturase 1 (SCD1), elongation of long-chain fatty acids family member 6 (ELOVL6), and acetyl CoA carboxylase (ACC). Par ailleurs, le glucose peut entrer dans les hépatocytes via glucose transporter 2 (GLUT2) dans les hépatocytes et entrer dans la voie de la glycolyse. Le glucose est phosphorylé en glucose 6-phosphate (G6P) suivi par son isomerisation en fructose 6-phosphate (F6P) et par la suite phosphorylé en fructose-2,6-bisphosphate (F2,6P). G6P et F2,6P peuvent conduire à la dephosphorylation du carbohydrate response element binding protein (ChREBP) et la dissociation de la protéine 14-3-3. Ceci permet la localisation nucléaire du ChREBP et la transcription de ses gènes cibles de la LDN dont FAS, SCD1, ELVL6, ACC et le gène de la pyruvate kinase L/R (Pklr). PPARγ permet l’activation de différents gènes impliqués dans le transport des acides gras (AG) et dans la biosynthèse des triglycérides (TG) favorisant ainsi la LDN et la mise en place d’une stéatose hépatique. © Adapté de (Sanders et Griffin, 2016).
Les gouttelettes lipidiques sont considérées comme une organelle très dynamique qui possède une machinerie cellulaire spécifique permettant la régulation de chacun de leur aspect biologique. Les gouttelettes lipidiques sont composées d’un noyau de lipides neutres entouré d’une monocouche phospholipidique et de protéines spécifiques qui se lient à leurs surfaces. Dans les hépatocytes, la taille des gouttelettes lipidiques peut varier de 300 à 800 nm pour les plus petites et aller jusqu’à quelques dizaines de microns de diamètre pour les plus importantes. Les lipides neutres constituant les gouttelettes lipidiques sont représentés principalement par les triglycérides, et sont synthétisés par des enzymes qui résident essentiellement dans le RE. La synthèse des triglycérides, voir figure 6, est catalysée par la famille des GPAT (glycerol-3-phosphate acyltranférase) qui réside à la fois dans le réticulum endoplasmique (RE) et dans la mitochondrie et qui catalyse la première étape de la synthèse des triglycérides pour aboutir à la formation de l’acide lysophosphatidique (LPA), aussi connu sous le nom de 1-acylglycérol-3-phosphate. L’ajout d’un deuxième acide gras au LPA est assuré par les enzymes de la famille des AGPAT (1-acylglycerol-3-phosphate acyltransferase) et génère de l’acide phosphatidique (PA).
Le acide phosphatidique peut ensuite conduire à la formation de diacylglycérol (DAG) par l’action de protéines de la famille des lipins qui ont une activité enzymatique PAP (phosphatidate phosphatase-1). Enfin, le DAG est converti en triacylglycérol sous l’action des DGAT (diacylglycerol O-acyltransferase). Il semblerait que la formation des gouttelettes lipidiques soit due à une accumulation de lipides neutres dans la bicouche du RE en raison de leur synthèse dans ce compartiment. Une fois un seuil critique atteint, un bourgeonnement vers le cytoplasme s’effectue, et durant ce processus les gouttelettes lipidiques naissantes acquièrent un certain nombre de protéines spécifiques, comme les périlipines (Plin). Les gouttelettes lipidiques naissantes peuvent atteindre une taille plus grande par relocalisation des enzymes spécifiques de la synthèse des triglycérides, du RE vers le site de la gouttelette lipidique en formation. Parmi ces enzymes nous trouvons la GPAT4 (glycerol-3-phosphate acyltransferase 4) et DGAT qui catalysent respectivement la première et dernière étape de la synthèse des triglycérides.
C. L’export des triglycérides par VLDL : Les triglycérides ont besoin d’être incorporés dans des very low density lipoproteins (VLDL) pour être sécrétés dans la circulation sanguine et être délivrés aux muscles, afin d’y être oxydés et fournir de l’énergie, ou aux tissus adipeux pour y être stockés. La structure des VLDL, présentée figure 8, possède un coeur constitué de triglycérides et de cholestérols, entouré de phospholipides et d’apolipoprotéines.
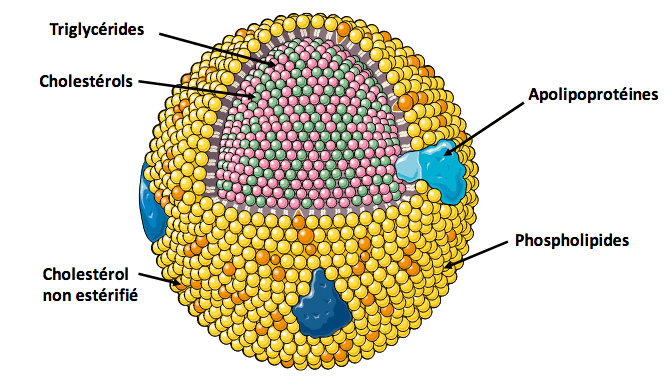 Figure 8 : Schéma de la structure d’une VLDL.
Figure 8 : Schéma de la structure d’une VLDL.
La formation des VLDL, présentée Figure 9, se déroule en deux étapes clés dont la première se déroule dans la lumière du réticulum endoplasmique rugueux (RER). Cette première étape, implique la microsomal triglyceride transfer protein (MTP), qui va agir en incorporant une petite quantité de triglycérides avec l’apolipoprotéine B (apoB) 100, directement après sa traduction par les ribosomes et son transfert à travers la membrane du RER. Le complexe intermédiaire ainsi formé est appelé VLDL naissante. La protein disulfide isomerase (PDI) est une sous unité de MTP, qui est nécessaire à son bon fonctionnement. De plus, il est à noter que l’ApoB existe sous 2 formes chez l’homme, la forme longue appelé apoB100 qui fait 550 kDa et la forme tronquée apoB48 de 210 kDa correspondant à la forme N-terminal de l’apoB100.
Avec une copie par particule, l’apoB100 constitue une protéine essentielle à la formation et à la sécrétion des VLDL et LDL. En revanche, l’apoB48 constitue la principale apolipoprotéine des chylomicrons sécrétés par l’intestin, pour permettre le transport des acides gras après un repas. Cette VLDL naissante est également recouverte de phospholipides, favorisé par la phospholipid transfert protein (PLTP). Lors de la deuxième étape, connue sous le nom de lipidation, des triglycérides supplémentaires vont fusionner avec la VLDL naissante, potentiellement dans le RE lisse (REL). Cette étape dépend de la disponibilité des triglycérides, et dans le cas d’une absence de lipidation, une quantité importante de VLDL naissantes est dégradée par le protéasome. Par ailleurs, la localisation de cette étape de lipidation n’est pas encore clairement définie et pourrait se dérouler au sein d’un ou plusieurs compartiments tel que le RE, le cytosol ou encore l’appareil de Golgi. Ce processus de lipidation semble également engager l’activité intracellulaire de l’apoC3, qui mobilise les triglycérides dans le RE pour la fusion avec les particules de VLDL nouvellement formées. Un prérequis à la sécrétion des VLDL par les hépatocytes est leur passage par le cis-Golgi. En effet, celui-ci fournit les messages d’adressage d’exocytose et permet ainsi la formation des VLDL matures.
Le transport des VLDL du RE vers le cis-Golgi se fait grâce à une vésicule spécialisée, la vésicule de transport des VLDL (VTV, VLDL transport vesicle). La formation de ces vésicules se fait par bourgeonnement à partir de la membrane du RE, et requiert, au moins, 5 protéines (Sar1, Sec23, Sec24, Sec13 et Sec31) regroupées sous le nom de coat complex II (COPII) ainsi que la protéine DFF45-like effector B (CideB). Cette VTV permet le transport des VLDL naissantes jusqu’à l’appareil de Golgi et va fusionner avec pour délivrer ces VLDL. Une fois délivrées, les VLDL naissantes subissent des modifications importantes avec l’acquisition de nouvelles apolipoprotéines et modifications post-traductionnelles qui permettent la formation de VLDL matures pouvant être transportées jusqu’à la membrane plasmique par une post- Golgi VLDL transport vesicle (PG-VTV) pour être exocytées.
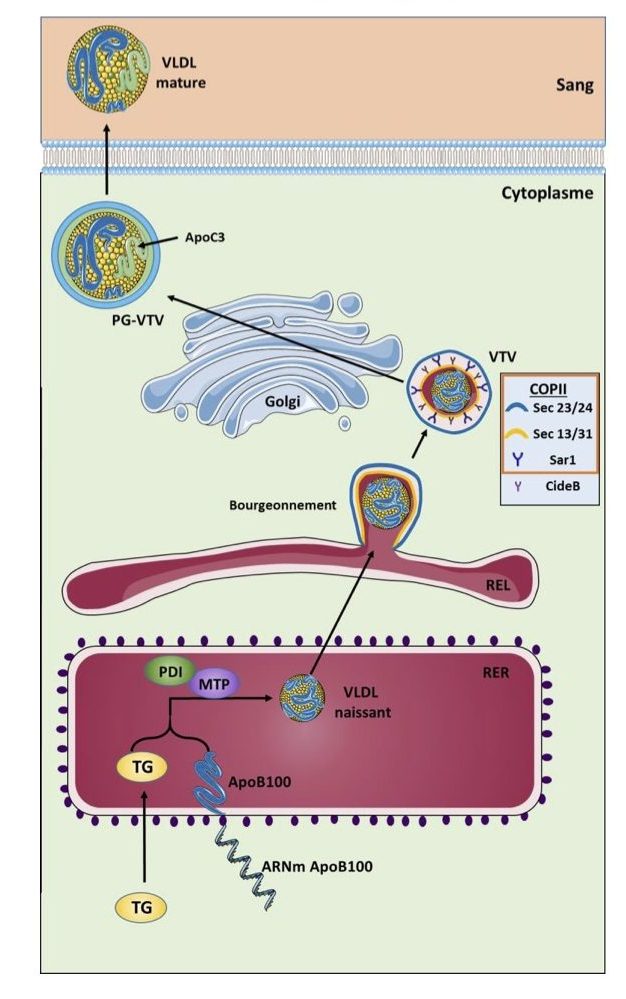 Figure 9. Schéma de la biogenèse des VLDL. Les triglycérides (TG) nouvellement synthétisés sont utilisés pour la formation et la lipidation des very low density lipoproteine (VLDL) initiées dans la lumière du réticulum endoplasmique rugueux (RER). La première étape est médiée par la microsomal triglyceride transfer protein (MTP), qui facilite l’association des TG avec l’apolipoprotéine B 100 (ApoB100) fraîchement traduit par les ribosomes pour former une VLDL naissante. Cette VLDL naissante passe ensuite dans le RE lisse (REL) où d’autres TG peuvent s’ajouter durant le processus de lipidation et également durant le bourgeonnement qui va permettre la formation d’une vésicule de transport des VLDL (VTV). Le CideB (DFF45-like effector B) est localisé sur la membrane du RE et VTV. CideB se lie au VLDL naissant et facilite la formation du VTV, pour cela il interagit avec Sar1 et les Sec (13, 23, 24, 31) pour assembler une couche complexe de COPII (Coat complex II), qui est nécessaire, notamment pour les vésicules de grande taille à partir du REL. Le VTV permet le transport jusqu’au cis-Golgi dans lequel la VLDL est libérée et va poursuivre sa maturation et lipidation. La VLDL est ensuite transportée jusqu’à la membrane plasmique pour être sécrétée. Ce transport est médié par la post-golgi VLDL transport vesicle (PG-VTV). © Adapté de (Alves-Bezerra et Cohen, 2017; Hossain et al., 2014; Olofsson et al., 2000; Tiwari et al., 2013; Tiwari et Siddiqi, 2012).
Figure 9. Schéma de la biogenèse des VLDL. Les triglycérides (TG) nouvellement synthétisés sont utilisés pour la formation et la lipidation des very low density lipoproteine (VLDL) initiées dans la lumière du réticulum endoplasmique rugueux (RER). La première étape est médiée par la microsomal triglyceride transfer protein (MTP), qui facilite l’association des TG avec l’apolipoprotéine B 100 (ApoB100) fraîchement traduit par les ribosomes pour former une VLDL naissante. Cette VLDL naissante passe ensuite dans le RE lisse (REL) où d’autres TG peuvent s’ajouter durant le processus de lipidation et également durant le bourgeonnement qui va permettre la formation d’une vésicule de transport des VLDL (VTV). Le CideB (DFF45-like effector B) est localisé sur la membrane du RE et VTV. CideB se lie au VLDL naissant et facilite la formation du VTV, pour cela il interagit avec Sar1 et les Sec (13, 23, 24, 31) pour assembler une couche complexe de COPII (Coat complex II), qui est nécessaire, notamment pour les vésicules de grande taille à partir du REL. Le VTV permet le transport jusqu’au cis-Golgi dans lequel la VLDL est libérée et va poursuivre sa maturation et lipidation. La VLDL est ensuite transportée jusqu’à la membrane plasmique pour être sécrétée. Ce transport est médié par la post-golgi VLDL transport vesicle (PG-VTV). © Adapté de (Alves-Bezerra et Cohen, 2017; Hossain et al., 2014; Olofsson et al., 2000; Tiwari et al., 2013; Tiwari et Siddiqi, 2012).
Une réduction de la sécrétion des triglycérides par les VLDL peut conduire à une accumulation de triglycérides. Cette réduction peut subvenir à la suite d’un manque génétique en ApoB et MTP, qui va conduire respectivement à une hypobêtalipoprotéinémie et une abêtalipoprotéinémie. De plus, des mutations rares sur ces protéines peuvent être associées avec des maladies hépatiques. La régulation de la sécrétion des VLDL ne semble pas encore totalement élucidée mais semble impliquer la disponibilité en triglycérides, l’apoB et d’autre apolipoprotéines comme apoC3, et certains mécanismes de transduction du signal. En effet, la voie des VLDL est étroitement liée à la présence de triglycérides, habituellement apportés par les chylomicrons après un repas ou produit par la LDN via le glucose. Ceci permet, physiologiquement, d’avoir une sécrétion constante des VLDL, afin d’alimenter les muscles en énergie. Un défaut de la LDN aurait des conséquences sur la sécrétion des VLDL, avec certaines protéines de la LDN décrites comme importantes pour la production des VLDL, tels que SCD1 ou DGAT2. Par ailleurs, il a été montré que l’expression des gènes MTTP et APOC3 est régulée négativement par l’insuline, principalement via Forkhead box protein O1 (FoxO1) et son exclusion du noyau, mais également via la voie Mitogen-activated protein kinases (MAPK). De plus, l’expression de l’apoB semblerait également négativement régulée par une forte dose d’insuline, potentiellement via activation de la voie PI3K ou inhibition de PTEN. L’activation de FXR semblerait jouer un rôle dans l’ inhibition de la sécrétion des VLDL.
La formation des triglycérides étant initialement réalisée dans le RE et potentiellement médiée par certaines de ses protéines chaperonnes, telles que BIP ou PDI ; un défaut ou stress de celui-ci peut avoir comme conséquence une diminution de la sécrétion des triglycérides par les VLDL. Le réticulum endoplasmique (RE) est l’un des organites les plus importants dans les cellules. Il joue un rôle majeur dans la synthèse, le repliement et la maturation structurelle de plus d’un tiers de toutes les protéines produites dans la cellule. En particulier, presque toutes les protéines destinées à résider dans le RE, la membrane plasmique, l’appareil de Golgi et les lysosomes. Des processus enzymatiques garantissent que les protéines sécrétées soient correctement repliées, modifiées et assemblées en complexes multiprotéiques dans le RE avant de pouvoir circuler plus en aval dans la voie de sécrétion. Le taux de succès d’un repliement correct reste cependant assez faible (moins de 20%) pour de nombreuses protéines transférées dans le RE. Comme les protéines de la voie de sécrétion jouent souvent un rôle crucial dans la signalisation (récepteurs de la surface cellulaire, transporteurs ou hormones polypeptidiques), les formes incomplètement repliées ne sont pas tolérées par la cellule et sont plutôt éliminées selon un processus appelé dégradation associée à l’ER (ERAD, endoplasmic-reticulum-associated protein degradation). Processus pendant lequel les protéines mal formées (connu en anglais sous le terme de misfolded protein) sont extraites du cytosol pour être ensuite ubiquitinylées et dégradées par le protéasome.
Le terme de stress du RE correspond à une accumulation excessive et à l’agrégation de protéines mal formées dans la lumière du RE. Ceci peut être dû à une perte de l’homéostasie du RE, avec une réduction de son potentiel de repli des protéines qui peut être causé par différentes perturbations au sein de la cellule. Par chance, la cellule mesure constamment la quantité de protéines mal formées, et si cette quantité dépasse un seuil critique, il y a activation d’une voie de transduction du signal appelé unfolded protein response (UPR) qui va conduire à une réponse adaptative de la cellule par réduction du stress ou induction de voies pro-apoptotiques, présenté figure 10. L’UPR est initié par 3 protéines transmembranaires du RE : inositol-requiring enzyme 1α (IRE1α), protein kinase RNA-like endoplasmic reticulum kinase (PERK), et activating transcription factor 6 (ATF6). En conditions normales, la protéine chaperonne, binding immunoglobulin protein (BIP), aussi connue sous le nom glucose regulated proteine 78 (GRP78), est associée à ces protéines et les inactive. Cependant, en période de stress du RE, BIP reconnait et se fixe également aux protéines mal formées. BIP n’étant plus disponible en quantité suffisante pour inhiber les branches de l’UPR, celles-ci sont donc activées. Cette activation de l’UPR va avoir plusieurs fonctions : la réduction de l’augmentation des protéines malformées, qui est réalisée par une régulation positive des protéines chaperonnes et enzymes de repliement des protéines ainsi que par l’augmentation des protéines associées à l’ERAD. Un rétrocontrôle de l’UPR pour éviter une hyperactivation de celui-ci lorsque l’homéostasie sera rétablie. Elle permet également une régulation du devenir de la cellule, par un contrôle des signaux pro-apoptotique et anti-apoptotique.
IRE1α est la branche de l’UPR la mieux conservée dans l’évolution, son activation se réalise par autophosphorylation puis dimérisation, une fois séparée de la molécule chaperonne BiP. IRE1α, une fois activée, permet l’excision de 26 nucléotides de l’ARNm codant pour le facteur de transcription X- box-binding protein 1 (XBP1). La forme excisée de XBP1, appelé XBP1s (splice) à partir de XBP1u (unsplice), conduit à la forme active de XBP1 qui est par la suite transloquée dans le noyau pour induire l’expression de gènes codant pour des molécules chaperonnes (BiP) et protéine permettant le maintien de l’homéostasie du RE et de l’ERAD. La branche IRE1α/XBP1 peut réguler la LDN dans les hépatocytes en se fixant directement sur des promoteurs de gènes, tels que SCD1, DGAT2 et ACC, pour activer la lipogenèse. De plus, IRE1α possède un domaine RNase, qui permet de dégrader des ARNm situés à proximité du RE. Ce processus est connu sous le nom de IRE1α-dependent decay (RIDD), et il permet de réduire le niveau de nombreux ARNm qui ont un rôle dans le métabolisme du glucose, des lipides, et l’apoptose, parmi lesquelles on trouve ANGPTL3 (angiopoietine-like 3) dont l’inactivation pourrait réduire la sécrétion des VLDL. IRE1α activé permet aussi une interaction avec la protéine tumor necrosis factor receptor-associated protein 2 (TRAF-2) et peut former un complexe avec la protéine apoptosis signal-regulated kinase 1 (ASK1). Ce complexe peut activer c-Jun N terminal kinase (JNK) ce qui engendre la production d’EROs, activation de l’autophagie et de voies métaboliques de l’inflammation impliquant nuclear factor-κB (NF- κB).
PERK, comme pour IRE1α, s’autophosphoryle, puis se dimérise pour être sous sa forme active. Une fois activé PERK peut phosphoryler et activer l’α-subunit of eukaryotic initiation factor 2 (eIF2α), sur sa serine 51. Ceci entraîne une inhibition de la synthèse de protéine dans le RE pour prévenir l’accumulation de protéines malformées. De plus, cela entraine l’activation de facteur de transcription comme activating transcription factor 4 (ATF4). ATF4 peut ensuite être transloqué dans le noyau et permettre d’augmenter l’expression de gènes codant pour des protéines chaperonnes, tel que BiP, ainsi que des gènes impliqués dans l’autophagie, la biosynthèse d’acides aminées, la sécrétion des protéines et le facteur de transcription pro-apoptotique CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP).
L’activation de la troisième branche de l’UPR implique ATF6, qui une fois séparé de BiP est transloqué dans l’appareil de Golgi pour être clivé par des endopeptidase S1P et S2P, et permettre le relargage de la forme active de ATF6 qui va agir avec ou sans XBP1s pour réguler l’expression de gènes de l’UPR.
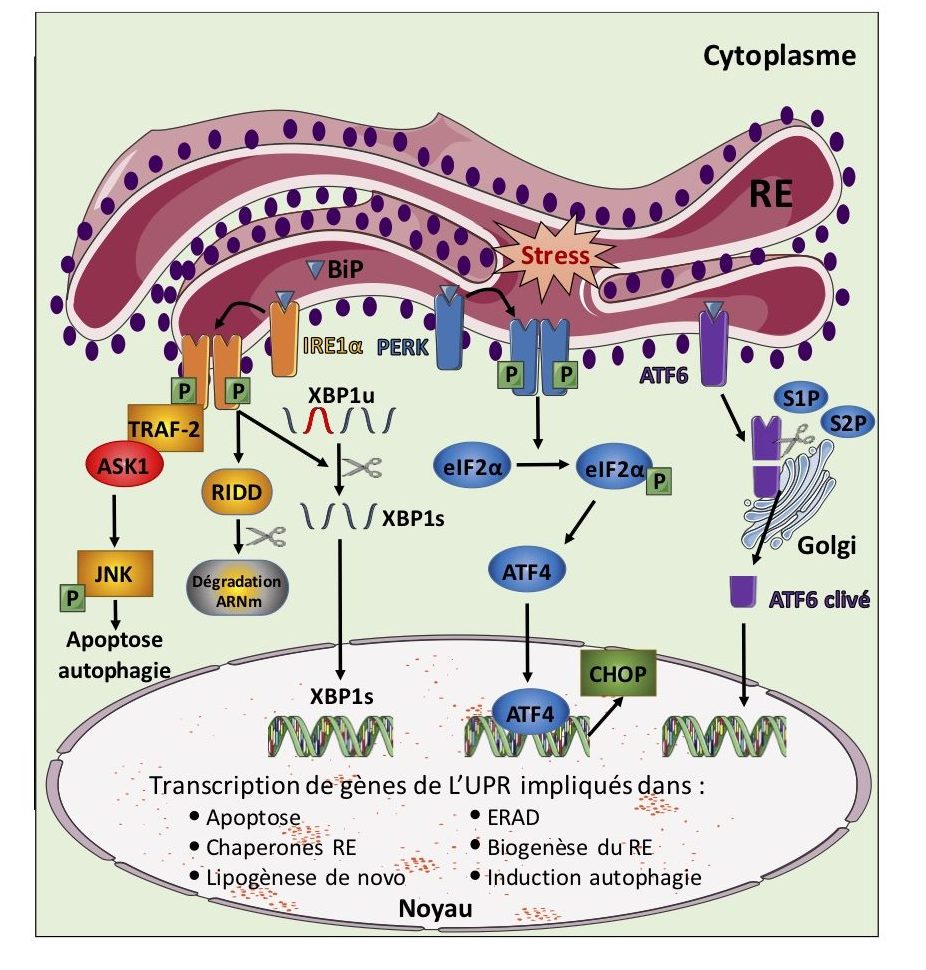 Figure 10. Les branches de l’UPR. Les trois senseurs de l’UPR (unfolded protein response), à savoir IRE1α (inositol-requiring enzyme 1α), PERK (protein kinase RNA-like endoplasmic reticulum kinase) et ATF6 (activating transcription factor 6), sont activés lorsque les agrégats de protéines mal repliées recrutent BiP (binding immunoglobulin protein) entraînant sa dissociation des senseurs. Cette dissociation a pour conséquence la dimérisation et la phosphorylation de IRE1α, cette activation induit l’épissage de l’ARNm de XBP1u (X-box-binding protein 1 unsplice) pour générer du XBP1s (splice) actif, qui joue un rôle dans l’augmentation de l’expression de gènes de l’UPR. IRE1α active également une autre voie cellulaire en intéragissant avec la kinase c-Jun N terminal (JNK), via le recrutement et activation de TRAF2 (tumor necrosis factor receptor-associated protein 2) et ASK1 (apoptosis signal-regulated kinase 1). PERK phosphoryle le facteur d’initiation de la traduction eIF2α (α-subunit of eukaryotic initiation factor 2), conduisant à l’atténuation globale des protéines et à l’activation de ATF4 (activating transcription factor 4), qui active l’expression de CHOP ((C/EBP) homologous protein). Dans des conditions de stress du RE, ATF6 est transporté vers l’appareil de Golgi, où son domaine cytosolique va cliver les protéases S1P et S2P, ce qui déclenche la transcription de protéine chaperonne du RE. Les facteurs de transcription XBP1s, ATF4 et ATF6 sont transloqués dans le noyau où ils activent l’expression des gènes cibles, tel que ER- associated-degradation (ERAD) ou des protéines chaperonnes. © Adapté de (Kabir et al., 2018).
Figure 10. Les branches de l’UPR. Les trois senseurs de l’UPR (unfolded protein response), à savoir IRE1α (inositol-requiring enzyme 1α), PERK (protein kinase RNA-like endoplasmic reticulum kinase) et ATF6 (activating transcription factor 6), sont activés lorsque les agrégats de protéines mal repliées recrutent BiP (binding immunoglobulin protein) entraînant sa dissociation des senseurs. Cette dissociation a pour conséquence la dimérisation et la phosphorylation de IRE1α, cette activation induit l’épissage de l’ARNm de XBP1u (X-box-binding protein 1 unsplice) pour générer du XBP1s (splice) actif, qui joue un rôle dans l’augmentation de l’expression de gènes de l’UPR. IRE1α active également une autre voie cellulaire en intéragissant avec la kinase c-Jun N terminal (JNK), via le recrutement et activation de TRAF2 (tumor necrosis factor receptor-associated protein 2) et ASK1 (apoptosis signal-regulated kinase 1). PERK phosphoryle le facteur d’initiation de la traduction eIF2α (α-subunit of eukaryotic initiation factor 2), conduisant à l’atténuation globale des protéines et à l’activation de ATF4 (activating transcription factor 4), qui active l’expression de CHOP ((C/EBP) homologous protein). Dans des conditions de stress du RE, ATF6 est transporté vers l’appareil de Golgi, où son domaine cytosolique va cliver les protéases S1P et S2P, ce qui déclenche la transcription de protéine chaperonne du RE. Les facteurs de transcription XBP1s, ATF4 et ATF6 sont transloqués dans le noyau où ils activent l’expression des gènes cibles, tel que ER- associated-degradation (ERAD) ou des protéines chaperonnes. © Adapté de (Kabir et al., 2018).
Le stress du RE est un mécanisme compliqué à étudier en raison des nombreuses causes qui peuvent amener à sa mise en place et qui impliquent généralement une mauvaise conformation des protéines. Parmi ces causes on trouve des défauts de la glycosylation des protéines, l’inhibition du protéasome (lieu de dégradation des protéines mal formées), stress oxydant, liaison covalente entre les protéines du RE à des métabolites réactifs, activation de la MAPK (mitogen-activated protein kinase), dysfonctionnement sévère de la mitochondrie, augmentation de la concentration calcique dans le cytoplasme et l’inhibition de la pompe calcique du RE de la famille des Sarco-Endoplasmique Reticulum Calcium ATPases (SERCAs). Cette dernière permet le maintien d’une concentration élevée de calcium, au sein du RE, qui est nécessaire à la bonne maturation des protéines.
D. L’import des acides gras : Les AGLC se déplacent dans la circulation sanguine sous forme d’AG libres liés à l’albumine après leur libération par des adipocytes ou sous forme de triglycérides contenus dans les VLDL et les chylomicrons. Ces triglycérides en circulation, sont hydrolysés par la lipoprotéine lipase (LPL) en acides gras libres, puis transloqués dans la cellule. Il semblerait qu’une partie des AG libres, notamment les AGCC et AGCM, peuvent diffuser librement à travers la membrane plasmique des cellules. Il existe cependant des mécanismes actifs de transport des acides gras à l’intérieur de la cellule et plusieurs protéines ont été identifiées pour avoir ce rôle de transporteur. On trouve la fatty acid translocase cluster of differenciation 36 (FAT/CD36), la famille des fatty acid transport proteins (FATP), les fatty acid binding protein (FABP) et les cavéolines. Ces transporteurs sont majoritairement exprimés de manière ubiquitaire avec quelques variations selon les tissus.
Les FAT/CD36, peuvent être stockés dans la cellule sous forme de vésicules contenant de nombreux transporteurs et qui vont fusionner avec la membrane plasmique pour augmenter le nombre de transporteurs et faciliter l’entrée d’AGCL dans la cellule. La régulation de FAT/CD36 et de sa translocation à la membrane est médiée par l’insuline et d’autres facteurs tel que PPARγ, pregnane X receptor (PXR) et liver X receptor (LXR). Les FATPs, constituent une famille de 6 transporteurs qui régulent l’entrée des AGCL et AGCTL. Il est à noter que FATP2 et FATP5 sont les principaux transporteurs exprimés dans le foie. Les FABPs comportent 9 isoformes parmi lesquels FABP1 et FABP5 sont les plus exprimés dans le foie. Les cavéolines sont au nombre de 3 protéines membranaires qui contribuent à la circulation des lipides et à la formation de gouttelettes lipidiques. La cavéoline 1 est particulièrement importante dans le foie et peut être impliquée dans certaines lésions métaboliques du foie.
E. L’autophagie : L’autophagie est un processus cellulaire qui permet la dégradation par le lysosome de constituants cytoplasmiques comme des protéines ou des organites. C’est un processus adaptatif de la cellule, en réponse à différentes formes de stress dont : la carence nutritionnelle, l’épuisement en facteur de croissance, les infections, le stress du RE ou l’hypoxie. La fonction principale de l’autophagie est de fournir les nutriments nécessaires au bon fonctionnement de la cellule pendant ces différentes formes de stress. L’autophagie permet également d’éliminer des composants cytosoliques de la cellule potentiellement dangereux et non désirés, comme des mitochondries endommagées ou des agrégats de protéines, pour protéger la cellule. Il existe plusieurs types d’autophagie : la microautophagie, l’autophagie médiée par des protéines chaperonnes et la plus commune, la macroautophagie. La macroautophagie se fait en plusieurs étapes avec en premier l’activation du complexe Unc-51- like kinase 1 (ULK1) en réponse à un stress qui déclenche ensuite la nucléation du phagophore par phosphorylation de composants du complexe de classe III PtdIns3K. Ces composants activent la production de PI3P (phosphatidylinositol-3-phosphate) au niveau du RE rugueux, ce qui permet le recrutement de protéines et complexes nécessaires à l’élongation du phagophore pour obtenir au final un autophagosome. Parmi ces protéines recrutées on trouve LC3-II (microtubule-associated protein light chain 3) et des protéines de la famille des ATG (autophagy related genes). Une fois l’autophagosome contenant les protéines et/ou organites est formé, celui-ci fusionne avec le lysosome pour former un autolysosome et permettre la dégradation du contenu de l’autophagosome, suivie de la libération des nutriments dans le cytoplasme.
2.2. Fonction épuratrice (détoxification) et de dégradation
Le foie a différente manière d’épurer et de dégrader les déchets entrant dans l’organisme ou sortant de celui-ci. Cette fonction d’épuration du foie est essentielle au bon fonctionnement du corps puisque cela permet une élimination des déchets qui s’ils s’accumulaient, pourraient nuire aux autres organes, comme par exemple le rein avec l’accumulation d’urée.
2.2.1 La production de bile : La bile est la sécrétion exocrine du foie. Son principal rôle est de favoriser l’absorption des graisses grâce aux sels biliaires. Chez l’homme, les hépatocytes secrètent quotidiennement environ 1L de bile. La bile est un liquide jaune (bile hépatique) ou vert olive (bile vésiculaire). Son pH est basique entre 7.6 et 8.6. Elle est principalement formée d’eau (97% pour la bile hépatique et 87% pour la bile vésiculaire) et d’acides biliaires (1.5 à 3% de la matière sèche de la bile), de phospholipides (appelés lécithines), de cholestérol (rendu soluble par les sels biliaires et la lécithine), de pigments biliaires (déchets provenant de la dégradation de l’hémoglobine et donnant sa couleur à la bile) et d’ions notamment de bicarbonates. En plus de son rôle crucial dans le métabolisme énergétique, le foie joue un rôle très important dans le métabolisme et l’élimination des xénobiotiques, et plus particulièrement des médicaments. En effet, après ingestion d’une substance et absorption gastro-intestinale, un premier passage hépatique est obligatoire avant d’atteindre la circulation sanguine. De plus, le foie possède un ensemble d’enzymes de métabolisme de ces xénobiotiques qui font de lui un organe capital dans la détoxification mais également une cible privilégiée lors de la formation de métabolites réactifs suite au métabolisme de certaines substances. C’est pourquoi le foie est sujet à de nombreuses études pharmacologiques et toxicologiques avec les différentes étapes du devenir du médicament qui regroupent l’absorption de la molécule, sa distribution dans l’organisme et son élimination, comprenant le métabolisme et l’excrétion.
La bile est sécrétée en continu par le foie, puis éventuellement stockée dans la vésicule biliaire qui la concentre ce qui explique une composition différente pour la bile hépatique et la bile vésiculaire (Tableau ci-dessous). La bile hépatique résulte à la fois de processus de sécrétion et d’excrétion. D’une part, les produits de sécrétion hépatique sont les phospholipides, les immunoglobulines A et les acides biliaires. Ces derniers, une fois parvenus dans l’intestin, peuvent se faire transformer par la flore bactérienne en acides biliaires secondaires capables de soit repasser dans le foie à partir de l’iléum (c’est le cycle entéro-hépatique) ou soit passer dans le sang. D’autre part, les produits d’excrétion se retrouvant dans la bile hépatique vont être le cholestérol, les pigments biliaires (bilirubine, …) et les métabolites des xénobiotiques. Ces fonctions d’excrétion sont assurées par des transporteurs membranaires (MDR, MRP…) situés sur les membranes des cellules bordant les canalicules biliaires.
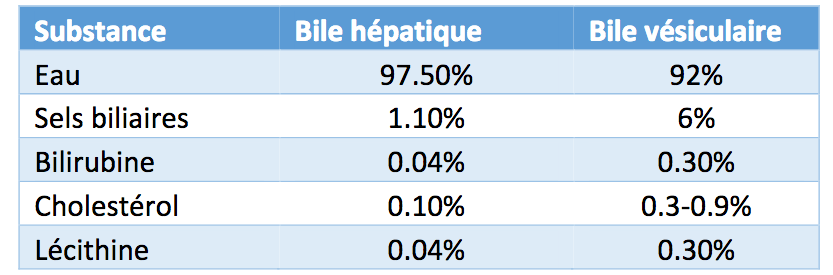
2.2.2. Métabolisme des xénobiotiques : Le foie étant placé entre le tube digestif et le reste de l’organisme, joue le rôle de «douane» pour contrôler lors de leur «premier passage», toutes les molécules absorbées. Les molécules faisant l’objet d’un premier passage sont généralement des xénobiotiques (molécules étrangères à l’organisme) mais également des substances physiologiques comme les sels biliaires par exemple. Certaines molécules très lipophiles, comme pour les lipides, échappent à l’effet de premier passage hépatique en passant par le circuit lymphatique comme c’est le cas pour les chylomicrons. La distribution d’un xénobiotique dépend généralement de l’origine de l’exposition. Lorsqu’un xénobiotique est ingéré, il y a un effet de premier passage hépatique avant d’être distribué dans la circulation sanguine et au reste de l’organisme. Tandis qu’une exposition par absorption pulmonaire d’un xénobiotique engendre une distribution à l’ensemble de l’organisme avant d’atteindre le foie et être métabolisé. De plus, les propriétés physico-chimiques du xénobiotique telle que sa masse molaire, sa solubilité ou son degré d’ionisation peuvent jouer sur les mécanismes d’absorption et de distribution de celui-ci. Par exemple, les molécules de petite taille ainsi que les composés lipophiles franchissent plus facilement les membranes, contrairement aux molécules ionisées.
Le métabolisme des xénobiotiques se déroule en plusieurs phases. La phase 0 correspond à l’étape d’entrée du xénobiotique dans la cellule qui peut se faire par diffusion simple ou par l’intermédiaire de transporteurs membranaires. S’ensuit une prise en charge par différentes enzymes du métabolisme des xénobiotiques qui sont catégorisées en 3 groupes : les enzymes de phase I, enzymes de phase II et les transporteurs membranaires qui sont des pompes à efflux pour exporter les molécules ou ses métabolites hors de la cellule. Les phases I et II permettent généralement de réaliser différentes réactions pour rendre les molécules plus faciles à éliminer de l’organisme et/ou moins actives. Les réactions de phase I consistent majoritairement à rendre le composé plus hydrophile et fournir des sites pour les réactions de conjugaison de la phase II, rendant le composé encore plus hydrophile. Les composés hydrophiles sont plus facilement exportés hors de la cellule par les pompes à efflux de la phase III, avant de se lier à des protéines de transport pour être éliminés via l’urine ou la bile.
A. Phase 0 : L’entrée des xénobiotiques et molécules endogènes dans les hépatocytes depuis la circulation sanguine se fait par le pôle baso-latéral. Elle peut être passive, facilitée ou active. Lors de cette dernière, l’entrée se fait par l’intermédiaire de transporteurs d’influx membranaires spécifiques de type solute carrier (SLC). Il existe différents types de ces transporteurs qui se différencient par la nature du xénobiotique qu’ils transportent. La NTCP (Na+-dependent taurocholate cotransporting polypeptide) assure le transport des acides biliaires et de certains médicaments (fluvastatine par exemple). La famille des OATP (organic anion transporting polypeptides) assure le transport d’acides organiques faibles de poids moléculaire supérieur à 300 g/mol, de certains médicaments zwitterioniques et peptides neutres linéaires et cycliques. Enfin, les OCTs (organic cation transporters) sont responsables du transport de cation, de carnitine et de certains médicaments comme le tramadole.
B. Phase I : La phase I correspond à une étape de fonctionnalisation des xénobiotiques, conduisant à la formation de métabolites. Dans le cas où ces métabolites obtiennent des propriétés hydrophiles suffisantes, ils peuvent directement être éliminés. Dans le cas contraire, le gain de sites de conjugaison va leur permettre d’augmenter leur caractère hydrophile grâce aux enzymes de phase II. Les différentes réactions catalysées par les enzymes de phase I sont l’oxydation, la réduction et l’hydrolyse. L’oxydation des xénobiotiques est la principale réaction, réalisée majoritairement par les cytochromes P450 (CYP) qui représentent plus des trois-quarts des enzymes de phase I. En plus des CYP on trouve parmi les enzymes de phase I, la flavine monooxygènase, l’alcool déshydrogénase, l’aldéhyde déshydrogénase, NADPH quinone oxydoréductase, époxydes hydrolase, estérases, monoamine oxydase et d’autres.
La phase I utilise donc principalement les CYP, et plus particulièrement ceux appartenant aux familles 1, 2 et 3. La monooxygénation est essentielle à la métabolisation de nombreux xénobiotiques, cependant dans certains cas, les CYP peuvent générer des métabolites réactifs plus toxiques que le substrat initial. En effet, un métabolite réactif possède une forte réactivité chimique qui lui permet de se fixer de manière covalente sur des macromolécules cellulaires, protéines ou acides nucléiques. Cette réactivité peut entraîner des effets délétères sur les fonctions cellulaires s’ils ne sont pas suffisamment pris en charge par les enzymes de phase II (époxyde-hydrolase, glutathion-S- transférases, UDP-glucuronosyltransférases).
C. Phase II : La phase II du métabolisme permet une biotransformation des composés endogènes et des xénobiotiques ayant subi la phase I. L’action des enzymes de phase II, appelé conjugaison, est de remplacer le groupement réactif (aldéhyde ou alcool) par un groupement moins réactif et rendant la molécule plus hydrophile pour ainsi faciliter son élimination. Les enzymes impliquées dans la phase II sont les UGTs (uridine 5′-diphospho-glucuronosyltransferases) qui assurent la glucuronidation d’un grand nombre de xénobiotiques et composés endogènes ; les SULTs (sulfotransferases) qui peuvent réaliser de la O-, N- ou S-sulfatation ; les GSTs (glutathione S-transferases) qui sont les principales enzymes de phase II. Les GSTs peuvent catalyser de nombreux types de réactions pour métaboliser les xénobiotiques et assurent également une défense primordiale contre les espèces réactives de l’oxygènes (EROs) ; NATs (N-acetyltransferases) qui catalysent des réactions d’acétylation et permettent la biotransformation de composés aromatiques.
D. Phase III : La phase III du métabolisme des xénobiotiques comprend des transporteurs actifs membranaires qui permettent l’efflux des métabolites hors des cellules pour qu’ils soient éliminés via la circulation sanguine puis les fèces, l’urine ou la bile. Les transporteurs ABC (ATP binding cassette carrier) impliqués dans ce processus comprennent 7 familles avec une vingtaine de transporteurs connus. Ces transporteurs sont répartis en deux groupes :
D.1. Les transporteurs de phase IIIa d’efflux ATP dépendant, qui sont localisés au niveau canaliculaire et assurent la sécrétion des xénobiotiques solubles et/ou de leurs métabolites dans la bile pour une élimination digestive. Cette élimination est assurée par BSEP (bile salt export pump), principalement connue pour assurer l’efflux dans la bile des acides biliaires ; P-gp (P-glycoprotein ou MDR1 pour multidrug-resistance proteins) qui peut transporter de nombreuses substances comme les médicaments cationiques amphiphiles volumineux, les hormones stéroïdes, les peptides hydrophobes et les glycolipides ; MRP2 (mutlidrug resistance-associated proteins) jouent également un rôle dans la sécrétion des acides biliaires ainsi que les métabolites conjugués à un glucuronide ou un sulfate ; BCRP (breast cancer resistance protein) connu pour conférer une résistance à des composés cytotoxiques tels que irinotecan, doxorubicine et d’autres médicaments comme le méthotrexate ; MATE (multidrug and toxin extrusion exchanger) qui utilise pour sa part un gradient électrochimique de cation pour assurer le transport dans les deux directions, et dont les substrats sont la créatinine, la guanidine et la thiamine, ainsi que de nombreux médicaments tels que la metformine, cimétidine, oxaliplatine et acyclovir. De plus MATE peut permettre l’entrée des médicaments cationiques et assurer l’excrétion biliaire de leurs métabolites.
D.2. Les transporteurs d’efflux de phase IIIb, sont des transporteurs sinusoïdaux qui assurent l’efflux de composés endogènes et xénobiotiques des hépatocytes vers la circulation sanguine pour permettre leur élimination par voie rénale. Parmi ces transporteurs on trouve MRP4 impliqué dans le transport de nombreux médicaments : antiviral, anticancéreux et cardiovasculaire, ainsi que des composés endogènes tels que les hormones stéroïdiennes, les prostaglandines et les acides biliaires ; OSTα/β (organic solute transporter) dont les substrats incluent les hormones stéroïdiennes, digoxin et des composés endogènes tels que les acides biliaires et prostaglandines ; MRP3 et MRP5 sont également 2 transporteurs de phase IIIb mais peu décrits.
Le premier passage hépatique évèle que le rôle de certaines protéines comme les cytochromes P450 se r. Le cytochrome P450 est un système complexe d’isoenzymes qui par des réactions d’oxydoréductions permettent la transformation d’un xénobiotique en un métabolite. On recense environ une trentaine de ces isoenzymes dans l’espèce humaine au niveau du foie mais aussi au niveau intestinal. Quatre isoenzymes sont impliquées dans le métabolisme d’environ 90% des médicaments couramment utilisés : ce sont les CYP 1A2, CYP 2C9, CYP 2D6 et CYP 3A4 ; les isoenzymes CYP 2B6, 2C8 et 2C19 n’étant impliquées que dans quelques interactions seulement. Ces isoenzymes seront donc sujettes à prendre en charge des substances qui peuvent soit les stimuler soit les inhiber, ce qui peut alors provoquer des interactions entre deux substances métabolisées par la même isoenzyme. Outre les cytochromes P450, d’autres enzymes hépatiques sont impliquées dans la détoxification de substances endogènes ou exogènes : elles sont regroupées sous le nom d’enzymes de phase 2. En effet, lors du captage d’un xénobiotique par un hépatocyte, celui-ci l’oxyde ou l’hydrolyse par l’intermédiaire des cytochromes afin de le rendre plus hydrophile et, lorsque cela n’est pas assez, d’autres enzymes peuvent rendre les métabolites encore plus hydrophiles afin de faciliter l’élimination. Ces enzymes sont principalement des transférases (glutathion-S-transférases, glucuronyltransférases, …), et permettent donc de catalyser des réactions dites de conjugaison.
2.2.3. Le cycle de l’urée : Le cycle de l’urée (CH₄N₂O, ou carbamide) permet l’élimination de l’ammoniac (NH₃) issu de la dégradation des groupements azotés des protéines (des acides aminés) et de la digestion d’aliments par la flore intestinale. Il se déroule au niveau hépatocytaire, plus précisément dans la mitochondrie et le cytosol de ces cellules. Ce cycle permet, à partir d’ammoniac (NH₃) et d’aspartate (C4H7NO4), de récupérer deux atomes d’azotes qui suite à une succession de 4 transformations permet la synthèse d’une molécule di-azotée CH₄N₂O appelée «urée» (ou carbamide) qui sera éliminée par le rein. Il est ainsi essentiel de retenir que tout dysfonctionnement du foie causé par exemple par l’alcoolisme ou par une inflammation hépatique altère le rôle de détoxification du foie, donc l’uréogenèse, ce qui entraînera une élévation de l’ammoniac dans la circulation sanguine. Le cerveau étant très sensible à l’hyperammoniémie, l’excès d’ammoniac peut provoquer une élévation excessive de deux neurotransmetteurs excitateurs : le glutamate et la glutamine qui, ayant une importante excitotoxicité, peuvent entraîner des complications neuronales (dont l’encéphalopathie hyperammoniémique) pouvant aller jusqu’au coma ou la mort. 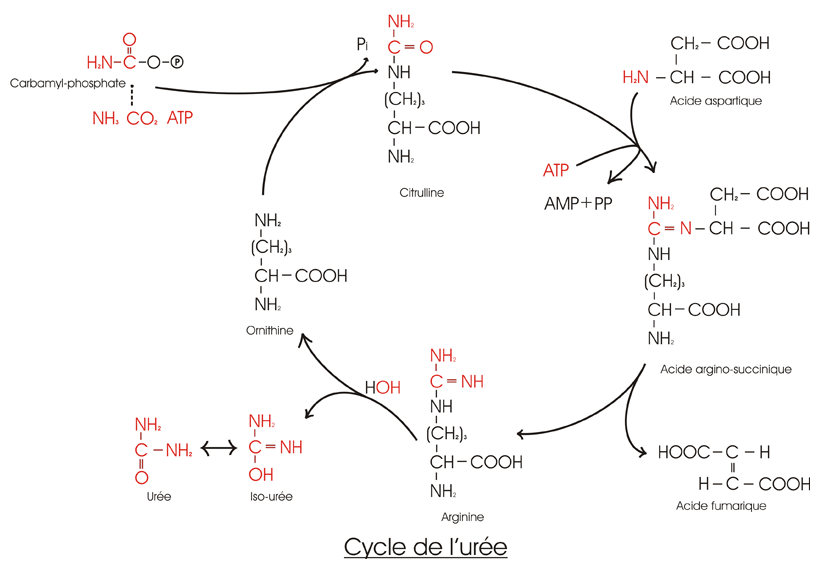 Figure 10. Cycle de l’urée. Dans la mitochondrie, une enzyme, la carbamyl phosphate synthétase I (CPS I), produit le carbamyl-phosphate à partir de l’ion ammonium (NH4+), de l’ATP et du dioxyde de carbone (CO2). Cette réaction se fait en deux étapes consommant chacune une molécule d’ATP. Le carbamyl apporte l’atome de carbone et un atome d’azote de la future molécule d’urée. La CPS I doit être activée par le N-acétylglutamate, effecteur allostérique. Chez certains animaux, dont les dipneustes, cette réaction est catalysée par une CPS III. (NB La CPS II est impliquée dans la synthèse des bases pyrimidiques, cette isoenzyme est cytosolique.) Le carbamyl-phosphate résultant se condense ensuite avec l’ornithine pour donner la citrulline. Cette réaction est catalysée par l’ornithine transcarbamylase (OTC). La citrulline est exportée dans le cytoplasme par diffusion facilitée. La citrulline se condense, en présence d’ATP, avec une molécule d’acide aspartique qui apporte le deuxième atome d’azote de l’urée. L’enzyme (arginosuccinate synthase AS) possède la particularité de produire de l’AMP à partir de l’ATP, clivage qui permet la libération d’une énergie de réaction plus importante pour la réaction grâce à la pyrophosphatase qui hydrolyse l’ion pyrophosphate alors formé. En effet, l’étude de l’énergétique de la réaction montre que l’hydrolyse classique de l’ATP en ADP n’apporterait pas suffisamment d’énergie pour la réaction, la variation d’énergie libre entre les substrats principaux et l’arginosuccinate étant trop élevée. Dans un second temps, l’AMP réagit avec une molécule d’ATP dans le cytoplasme pour régénérer de l’ADP : finalement deux ATP sont bel et bien consommés. Ensuite, l’acide arginosuccinique est converti en arginine et en acide fumarique par l’arginosuccinate lyase (AL). L’arginase catalyse l’hydrolyse de l’arginine en ornithine et urée en consommant une molécule d’eau. L’ornithine est ainsi régénérée et peut regagner la mitochondrie pour fixer une nouvelle molécule de carbamyl-phosphate.
Figure 10. Cycle de l’urée. Dans la mitochondrie, une enzyme, la carbamyl phosphate synthétase I (CPS I), produit le carbamyl-phosphate à partir de l’ion ammonium (NH4+), de l’ATP et du dioxyde de carbone (CO2). Cette réaction se fait en deux étapes consommant chacune une molécule d’ATP. Le carbamyl apporte l’atome de carbone et un atome d’azote de la future molécule d’urée. La CPS I doit être activée par le N-acétylglutamate, effecteur allostérique. Chez certains animaux, dont les dipneustes, cette réaction est catalysée par une CPS III. (NB La CPS II est impliquée dans la synthèse des bases pyrimidiques, cette isoenzyme est cytosolique.) Le carbamyl-phosphate résultant se condense ensuite avec l’ornithine pour donner la citrulline. Cette réaction est catalysée par l’ornithine transcarbamylase (OTC). La citrulline est exportée dans le cytoplasme par diffusion facilitée. La citrulline se condense, en présence d’ATP, avec une molécule d’acide aspartique qui apporte le deuxième atome d’azote de l’urée. L’enzyme (arginosuccinate synthase AS) possède la particularité de produire de l’AMP à partir de l’ATP, clivage qui permet la libération d’une énergie de réaction plus importante pour la réaction grâce à la pyrophosphatase qui hydrolyse l’ion pyrophosphate alors formé. En effet, l’étude de l’énergétique de la réaction montre que l’hydrolyse classique de l’ATP en ADP n’apporterait pas suffisamment d’énergie pour la réaction, la variation d’énergie libre entre les substrats principaux et l’arginosuccinate étant trop élevée. Dans un second temps, l’AMP réagit avec une molécule d’ATP dans le cytoplasme pour régénérer de l’ADP : finalement deux ATP sont bel et bien consommés. Ensuite, l’acide arginosuccinique est converti en arginine et en acide fumarique par l’arginosuccinate lyase (AL). L’arginase catalyse l’hydrolyse de l’arginine en ornithine et urée en consommant une molécule d’eau. L’ornithine est ainsi régénérée et peut regagner la mitochondrie pour fixer une nouvelle molécule de carbamyl-phosphate.
2.2.4. Epuration vasculaire de la bilirubine : Le foie exerce également une activité dans l’épuration des globules rouges et de leur hémoglobine. En effet, les érythrocytes ont une durée de vie d’environ 120 jours et sont détruits dans la rate, où la dégradation de l’hémoglobine produit successivement de la biliverdine puis de la bilirubine libre. Le fer contenu dans l’hème est recyclé et est restocké dans les hépatocytes au niveau de la ferritine afin d’être remobilisé selon les besoins. La bilirubine libre est toxique et peut être nocive ; elle possède une couleur jaune caractéristique. Elle parvient au foie par voie sanguine grâce au transport par l’albumine et y est transformée par une enzyme hépatique, la glucuronyltransférase, en un produit non toxique : la bilirubine conjuguée. Celle-ci est ensuite déversée dans la bile, dont elle est un des composants majeurs. La bile, éliminée par les intestins, voit sa bilirubine conjuguée transformée par les bactéries de la flore intestinale en un pigment incolore qui est l’urobilinogène. Lors de l’élimination fécale de la bile, l’urobilinogène se fait à son tour transformer en pigment marron/noir toujours par la flore intestinale : c’est le stercobilinogène, qui une fois oxydé en stercobiline, donne aux selles sa couleur brune. Une partie de la bile se faisant éliminer, le reste de la bile va soit utiliser le cycle entérohépatique pour revenir au niveau du foie, soit passer dans le compartiment systémique, ce qui aura un impact différent sur la métabolisation de la bilirubine conjuguée. La bile sera alors éliminée par les reins qui par oxydation va transformer l’urobilinogène en urobiline qui est le pigment jaune donnant aux urines sa couleur. La capacité de la bilirubine à pouvoir se pigmenter selon son métabolisme est une chose importante car elle peut vite marquer un dysfonctionnement hépatique lorsqu’un ictère se déclenche chez une personne.
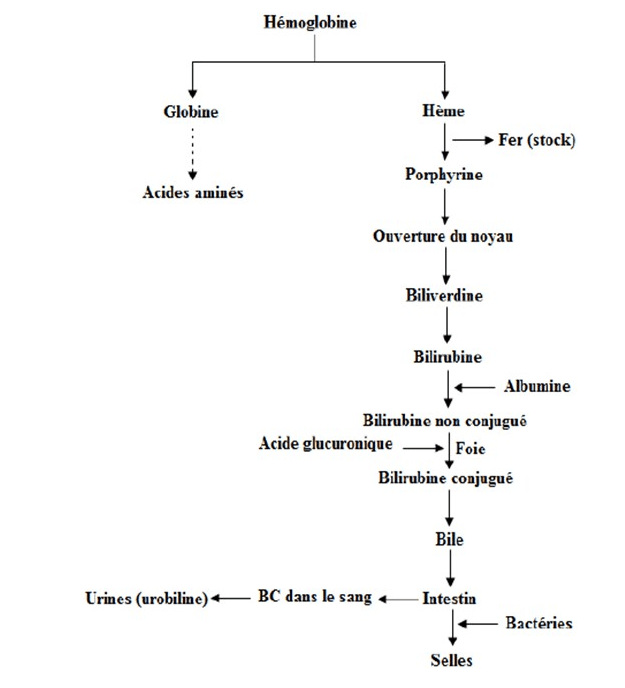 Figure 11. Dégradation physiologique de l’hémoglobine. Les érythrocytes parvenus en fin de vie du fait de leur sénescence ou de leur altération sont retirés du sang par phagocytose par des macrophages dans la rate et dans le foie. En cas d’hémolyse dans la circulation sanguine, l’hémoglobine se lie à l’haptoglobine, tandis que l’hème libre est fixé par l’hémopexine, ce qui limite l’effet oxydant. L’hémoglobine incomplètement dégradée ou libérée en trop grande quantité à partir de globules rouges endommagés est susceptible d’obstruer les vaisseaux sanguins, notamment les capillaires des reins, ce qui peut provoquer des néphropathies. L’hémoglobine libérée est éliminée du sang par la protéine CD163, exclusivement exprimée dans les monocytes et les macrophages. L’hémoglobine est dégradée dans ces cellules et le fer de l’hème est recyclé, tandis qu’une molécule de monoxyde de carbone est libérée par molécule d’hème dégradée : la dégradation de l’hème est l’un des rares processus naturels produisant du monoxyde de carbone dans le corps humain et est responsable de la présence de CO dans le sang d’individus respirant même l’air le plus pur. Ce processus forme de la biliverdine, puis de la bilirubine, de couleur jaune. Insoluble, elle est libérée par les macrophages dans le plasma sanguin, où elle se lie à la sérum albumine, qui la transporte jusqu’aux hépatocytes. Ces derniers la solubilisent par conjugaison avec l’acide glucuronique et la sécrètent dans les intestins avec la bile. Les intestins métabolisent la bilirubine en urobilinogène, qui est excrété dans les fèces sous forme de stercobiline ainsi que dans les urines. Lorsque la bilirubine ne peut être excrétée, sa concentration sanguine augmente et elle est éliminée essentiellement par les urines, qui deviennent foncées tandis que les fèces sont décolorées. Le fer issu de la dégradation de l’hème est stocké dans les ferritines des tissus et véhiculé dans le plasma sanguin par des β-globulines telles que les transferrines.
Figure 11. Dégradation physiologique de l’hémoglobine. Les érythrocytes parvenus en fin de vie du fait de leur sénescence ou de leur altération sont retirés du sang par phagocytose par des macrophages dans la rate et dans le foie. En cas d’hémolyse dans la circulation sanguine, l’hémoglobine se lie à l’haptoglobine, tandis que l’hème libre est fixé par l’hémopexine, ce qui limite l’effet oxydant. L’hémoglobine incomplètement dégradée ou libérée en trop grande quantité à partir de globules rouges endommagés est susceptible d’obstruer les vaisseaux sanguins, notamment les capillaires des reins, ce qui peut provoquer des néphropathies. L’hémoglobine libérée est éliminée du sang par la protéine CD163, exclusivement exprimée dans les monocytes et les macrophages. L’hémoglobine est dégradée dans ces cellules et le fer de l’hème est recyclé, tandis qu’une molécule de monoxyde de carbone est libérée par molécule d’hème dégradée : la dégradation de l’hème est l’un des rares processus naturels produisant du monoxyde de carbone dans le corps humain et est responsable de la présence de CO dans le sang d’individus respirant même l’air le plus pur. Ce processus forme de la biliverdine, puis de la bilirubine, de couleur jaune. Insoluble, elle est libérée par les macrophages dans le plasma sanguin, où elle se lie à la sérum albumine, qui la transporte jusqu’aux hépatocytes. Ces derniers la solubilisent par conjugaison avec l’acide glucuronique et la sécrètent dans les intestins avec la bile. Les intestins métabolisent la bilirubine en urobilinogène, qui est excrété dans les fèces sous forme de stercobiline ainsi que dans les urines. Lorsque la bilirubine ne peut être excrétée, sa concentration sanguine augmente et elle est éliminée essentiellement par les urines, qui deviennent foncées tandis que les fèces sont décolorées. Le fer issu de la dégradation de l’hème est stocké dans les ferritines des tissus et véhiculé dans le plasma sanguin par des β-globulines telles que les transferrines.
2.3. Régulation hépatique de l’immunité
De par sa localisation et sa fonction, le foie est continuellement exposé aux antigènes alimentaires, aux antigènes provenant de la flore intestinale et à d’éventuels microorganismes pathogènes également. De plus, anatomiquement, la veine splénique est l’un des précurseurs de la veine porte, ce qui fait que les lymphocytes provenant de la rate passent par la veine porte pour ensuite traverser les sinusoïdes hépatiques afin d’atteindre la circulation systémique par l’intermédiaire de la veine centrolobulaire. Il s’avère que le faible flux sanguin et le diamètre réduit des sinusoïdes permettent aux lymphocytes T (en bleu sur la figure ci-dessous), aux cellules NK (en jaune) et aux cellules NKT (en vert) d’interagir avec l’épithélium vasculaire, les hépatocytes et les cellules de Küppfer (en violet). Celles-ci sont aussi en contact avec les antigènes, les toxines et les microorganismes provenant de l’intestin, permettant ainsi leur élimination.
Figure 12. Les différentes cellules immunitaires au sein des sinusoïdes hépatiques.
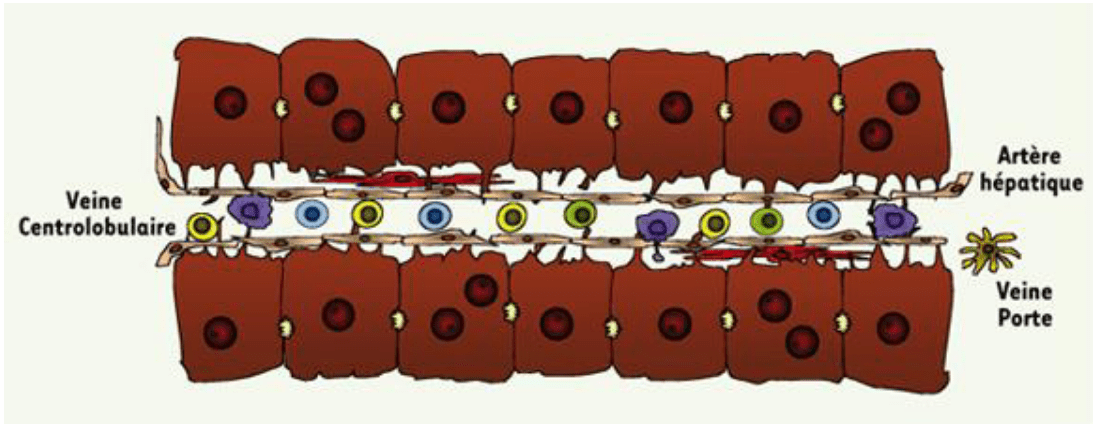 Les cellules NKT, présentes en grand nombre, ont la faculté de sécréter des cytokines de type Th1 et Th2 et ainsi d’orienter la réponse immune subséquente. Les cellules de Küppfer jouent le rôle de cellules présentatrices d’antigènes qui orchestrent également les premières phases de la réponse immune par l’intermédiaire de la sécrétion d’IL-12 et d’IL-18 ce qui déclenchent l’expansion et la différenciation des NK, cellules cytotoxiques sécrétant l’IFN-γ. Dans l’espace porte, loge également une population de cellules dendritiques hépatiques. Peu nombreuses et généralement immatures, ces cellules d’origine myéloïde sont aptes à phagocyter et à apprêter divers antigènes. La sécrétion constitutive d’IL-10 et de TGF-ß par les cellules de Küppfer et les cellules endothéliales sinusoïdales créent un microenvironnement rendant les cellules dendritiques tolérogéniques. L’architecture et la composition cellulaire particulière du foie, ainsi que les chimiokines et les cytokines qui y sont sécrétées, contribuent à faire du foie un milieu tolérogénique unique. Ainsi donc, on peut comprendre que lors d’une affection hépatique, un disfonctionnement de système immunitaire peut avoir lieu ce qui peut avoir un effet sur la capacité qu’a un individu à se protéger des micro-organismes pathogènes.
Les cellules NKT, présentes en grand nombre, ont la faculté de sécréter des cytokines de type Th1 et Th2 et ainsi d’orienter la réponse immune subséquente. Les cellules de Küppfer jouent le rôle de cellules présentatrices d’antigènes qui orchestrent également les premières phases de la réponse immune par l’intermédiaire de la sécrétion d’IL-12 et d’IL-18 ce qui déclenchent l’expansion et la différenciation des NK, cellules cytotoxiques sécrétant l’IFN-γ. Dans l’espace porte, loge également une population de cellules dendritiques hépatiques. Peu nombreuses et généralement immatures, ces cellules d’origine myéloïde sont aptes à phagocyter et à apprêter divers antigènes. La sécrétion constitutive d’IL-10 et de TGF-ß par les cellules de Küppfer et les cellules endothéliales sinusoïdales créent un microenvironnement rendant les cellules dendritiques tolérogéniques. L’architecture et la composition cellulaire particulière du foie, ainsi que les chimiokines et les cytokines qui y sont sécrétées, contribuent à faire du foie un milieu tolérogénique unique. Ainsi donc, on peut comprendre que lors d’une affection hépatique, un disfonctionnement de système immunitaire peut avoir lieu ce qui peut avoir un effet sur la capacité qu’a un individu à se protéger des micro-organismes pathogènes.
2.4. Régulation hormonale
Le foie a plusieurs fonctions principales qui sont associées aux hormones du corps. Par exemple :
2.4.1. le foie intervient dans la conversion chimique de l’hormone thyroïdienne (l’hormone T4) en sa forme active (l’hormone T3). Cette conversion chimique est réalisée par la 5’désiodase que l’on peut retrouver dans le foie mais également au niveau des intestins, du rein, du système nerveux central et de la thyroïde. Ainsi, bien qu’il y ait une régulation hormonale par les hépatocytes, le fait de retrouver cette enzyme dans d’autres organes fait qu’une affection hépatique a un impact minimal sur la régulation hormonale thyroïdienne.
2.4.2. Le foie sécrète, en réponse à la stimulation des hépatocytes par la Growth Hormone (GH), la IGF-1 (Insulin Growth Factor) qui est une hormone favorisant la croissance cellulaire. Outre le fait que l’hormone agisse sur le cartilage de conjugaison et a un rôle dans la régulation de la croissance d’un individu, cette hormone a également un impact au niveau cardiaque vu que la baisse d’IGF-1 a pu être liée à une augmentation de l’incidence des maladies cardiovasculaires.
2.4.3. L’angiotensinogène est une autre hormone fabriquée par le foie. Cette hormone est le précurseur du Système Rénine Angiotensine Aldostérone (SRAA) qui régule les taux de sodium et de potassium dans les reins et joue un rôle dans le contrôle de la pression artérielle. Dans ce même système intervient une enzyme grandement importante dans le contrôle de la pression artérielle, l’enzyme de conversion, qui est également produite par le foie. Celui-ci exerce donc un rôle essentiel dans la régulation de la tension artérielle en association avec le rein, les vaisseaux et le cœur.
2.5. La régénération hépatique
Sur un foie au repos, vu que les hépatocytes sont en phase G0 du cycle mitotique et sont donc quiescents, on observe très peu de mitose spontanée dans les hépatocytes. Cependant après hépatectomie ou lors d’une nécrose hépatique, chaque hépatocyte quiescent se fait stimuler par des cellules non-hépatiques, notamment des leucocytes, à travers une synthèse de facteurs inflammatoires (TNFα) et d’interleukines (IL-6, IL-7, IL-22) qui paradoxalement permettent de faire rentrer les hépatocytes dans le cycle mitotique. Cette stimulation permet alors à l’hépatocyte de se diviser 1 à 2 fois permettant ainsi la récupération de la masse hépatique. Ainsi, si on réalise une hépatectomie de 70% de la masse hépatique chez le rat, il y aura une récupération intégrale en 7 à 10 jours. Le rôle des hépatocytes dans la régénération hépatique est surtout admis dans les conditions physiologiques. En effet, dans des conditions pathologiques, l’utilisation de drogues inhibant la prolifération hépatocytes suivie d’une stimulation de régénération hépatique a permis de révéler l’existence de nouvelles populations cellulaires : les cellules ovales et les small hepatocytes like progenitor cells (SHPC).
2.5.1. Les cellules ovales : Les cellules ovales (cellules souches hépatiques), se trouvent au niveau du canal d’Herring et prolifèrent autour de l’espace porte avant de migrer vers le parenchyme hépatique. Leur présence révèle l’émergence d’un processus de régénération à partir de cellules précurseurs dans le foie humain, processus d’autant plus important que l’atteinte hépatique est sévère. Bien que sa présence chez l’Homme n’est pas encore été prouvée de manière concrète, elle l’a été chez le rat et la souris où plusieurs études probantes ont pu montrer que les cellules ovales jouaient un important rôle de régénération hépatique au cours des carcinomes hépatiques, des cholangiocarcinomes, des cirrhoses ainsi que des hémochromatoses. Comme toute cellule souche, les cellules ovales sont capables de se différencier en d’autres types de cellules autre que les hépatocytes. Ainsi, elles peuvent donner, lors de leur prolifération, des cellules biliaires, des cellules pancréatiques et des cellules de l’épithélium intestinal comme le montre le schéma ci-dessous. Certains mécanismes observés lors de la régénération hépatocytaire sont aussi retrouvés lors de la prolifération des cellules ovales. Ainsi, chez des souris déficientes en IL-6 et/ou de récepteur de type 1 du TNF-α, la prolifération des cellules ovales est fortement diminuée, ce qui montre qu’il existe une relation entre les processus régénératifs du tissu hépatique : les marqueurs inflammatoires, notamment certaines interleukines et les facteurs de nécroses tumorales permettent non seulement la stimulation mitotique des hépatocytes sains mais également d’orienter la différentiation des cellules ovales en de nouveaux hépatocytes.
Figure 13. La différenciation des cellules souches hépatiques.
2.5.2. Les SHPC : Les Small Hepatocyte-like progenitor cells (SHPC) sont, comme leur nom l’indique, des petites cellules ayant un rôle de progéniteur et étant morphologiquement semblables aux hépatocytes. En effet, elles expriment certains marqueurs hépatiques comme l’albumine et la transferrine par exemple et sont capables de synthétiser, en faible quantité de l’alpha-fœto protéine (AFP) qui est également produite par les cellules souches hépatiques (et les cellules ovales). Alors que certains chercheurs considéraient que les SHPC se trouvaient au niveau de l’espace porte, d’autres études réfutaient cette hypothèse en montrant qu’elles étaient localisées au niveau du parenchyme hépatique. L’activité des SHPC peut être considérée comme complémentaire à celle des cellules ovales lors de la synthèse d’hépatocytes dans le cadre d’une régénération hépatique. En effet, à la différence des hépatocytes et des cellules ovales, les SHPC expriment très faiblement les cytochromes P450 qui sont impliqués dans la détoxification hépatique comme vu plus haut. Ceci a été prouvé par une étude chez le rat lors de l’injection de rétrorsine qui est une molécule dont le métabolite pyrrolique produit par les cytochromes P450 désactive les protéines et acides nucléiques. Or, les SHPC n’exprimant que très peu de CYP, les études in-vitro ont montré que la transformation de la rétrorsine en son métabolite pyrrolique ne se faisait que très peu et qu’une très faible toxicité était observée vis-à-vis de ces cellules alors que l’apoptose était très importante dans les autres lignées des cellules hépatiques. Ceci permettant alors au foie de pouvoir continuer à synthétiser un pool d’hépatocytes afin de se régénérer lorsque les cellules ovales sont inhibées. Cependant, bien que les cellules ovales et les SHPC sont morphologiquement différents, elles répondent aux mêmes stimuli, c’est-à-dire aux IL-6 et TNF-α, montrant encore une fois le rôle fondamental du système immunitaire dans la stimulation et la différenciation des cellules progénitrices en hépatocytes lors d’agressions hépatiques.
2.6. Fonction de synthèse, régulation et dégradation protéique
Les hépatocytes ont un rôle essentiel dans la synthèse de protéines sanguines. En effet, le foie est capable de produire des protéines ou facteurs de coagulation, des protéines plasmatiques de transport et de l’inflammation ou encore des protéines rentrant dans le métabolisme du fer.
2.6.1. Protéines ou facteurs de coagulation : Tous les facteurs de la coagulation ont une synthèse exclusivement hépatique sauf le facteur VIII qui peut être produit par d’autres organes comme le rein par exemple. Il est très fréquent de voir des anomalies de la coagulation (hémorragies, …) dans une pathologie hépatique. Il existe deux voies de la coagulation : une voie extrinsèque, la plus rapide, et une voie intrinsèque. Chacune de ces voies utilisent ses propres facteurs mais bien que tous ces facteurs soient synthétisés par le foie, c’est le facteur V, facteur retrouvé dans la voie intrinsèque, qui est le facteur le plus représentatif de l’intégrité hépatique. Il est important de noter que le foie synthétise également les inhibiteurs de la coagulation comme la protéine C et la protéine S par exemple ou encore l’antithrombine, ce qui peut entraîner des troubles thrombotiques dans d’autres hépatopathies comme dans la cirrhose.
2.6.2. Protéines plasmatiques de transport : Les principales protéines de transport fabriquées par le foie sont les albumines qui sont des protéines globulaires de haut poids moléculaire (65-70 kDa). Ces albumines ont la capacité de se lier avec de nombreuses molécules (xénobiotiques, protéines, hormones, …) afin de les amener d’un endroit à l’autre de l’organisme. C’est ainsi que les albumines jouent un rôle primordial dans le transport et la dégradation par le foie de la bilirubine qui in fine donnera la couleur des urines et des selles (une couleur anormale de celles-ci montrant le plus souvent une défaillance hépatique). Les albumines jouent également un rôle essentiel dans la régulation de la pression osmotique et oncotique du sang et contrôle donc les entrées et sorties de liquides au sein du compartiment vasculaire. Physiologiquement, comme les pores du glomérule rénal ne sont pas assez grands pour les filtrer, les albumines ne sont pas éliminées dans les urines. Cependant, en présence d’anomalies de la barrière de filtration (ou éventuellement d’anomalies de réabsorption tubulaire), une quantité plus importante d’albumines et de protéines peut être retrouvée dans les urines, ce qui signe un état pathologique. Selon les concentrations d’albumines retrouvées dans les urines (albuminurie), on pourra parler de microalbuminurie ou de macroalbuminurie.
2.6.3. Protéines plasmatiques de l’inflammation : On appelle protéine de la réaction inflammatoire une protéine dont la concentration plasmatique varie d’au moins 25% durant la première semaine de l’inflammation. C’est le foie qui est en charge de la synthèse d’une partie de ces protéines comme par exemple la protéine C-réactive, le serum amyloïde A, l’haptoglobine, l’orosomucoïde, l’α2-macroglobuline ou encore l’α1-antitrypsine. Leur origine hépatique explique que ces marqueurs peuvent ne pas être augmentés en cas d’insuffisance hépatocellulaire.
2.6.4. Protéines impliquées dans le métabolisme du fer : Le fer est un métal essentiel notamment dans le fonctionnement enzymatique de l’organisme car il agit souvent en tant que cofacteur. Le fer est principalement stocké dans le foie par la ferritine qui est une protéine synthétisée par les hépatocytes. Ce fer peut se libérer de la ferritine afin de se répandre à travers tout le reste du corps en sortant des hépatocytes par un canal spécifique nommé la ferroportine et en étant transporté par une autre protéine produite par le foie : la transferrine. A l’instar des protéines de la coagulation, le foie produit également des protéines régulant négativement le métabolisme du fer à travers la synthèse d’hepcidine qui permet d’inhiber la sortie du fer hors des hépatocytes lorsque la ferritinémie est normale ou élevée. Ainsi, en cas d’hépatopathies, le métabolisme du fer pourra se retrouver totalement bouleversé à travers les troubles de synthèse de l’hepcidine, de la ferritine et de la transferrine, ce qui entraînera souvent des augmentations du bilan hépatique comme c’est le cas dans une cirrhose non compliqué.
Références
- Agius, L. (2008) Glucokinase and molecular aspects of liver glycogen metabolism. Biochemical Journal, vol. 414, n°1, p. 1‐18.
- Vekemans, K. et Braet, F. (2005) Structural and functional aspects of the liver and liver sinusoidal cells in relation to colon carcinoma metastasis. World Journal of Gastroenterology : WJG, vol. 11, n°33, p. 5095‐5102.
- Rmilah, A. A., Zhou, W., Nelson, E., Lin, L., Amiot, B. et Nyberg, S. L. (2019) Understanding the marvels behind liver regeneration. Wiley interdisciplinary reviews. Developmental biology, vol. 8, n°3, p. e340.
- Kietzmann, T. (2017) Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biology, vol. 11, p. 622‐630.
- Rui, L. (2014) “Energy Metabolism in the Liver”. In R. Terjung (dir.), Comprehensive Physiology (p. 177‐197). Hoboken, NJ, USA : John Wiley & Sons, Inc.
- Dikic, I. et Elazar, Z. (2018) Mechanism and medical implications of mammalian autophagy. Nature Reviews Molecular Cell Biology, vol. 19, n°6, p. 349‐364.
- Parzych, K. R. et Klionsky, D. J. (2014) An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxidants & Redox Signaling, vol. 20, n°3, p. 460‐473.
- Fernandez, M. A., Albor, C., Ingelmo-Torres, M., Nixon, S. J., Ferguson, C., Kurzchalia, T., Tebar, F., Enrich, C., Parton, R. G. et Pol, A. (2006) Caveolin-1 Is Essential for Liver Regeneration. Science, vol. 313, n°5793, p. 1628‐1632.
- Ipsen, D. H., Lykkesfeldt, J. et Tveden-Nyborg, P. (2018) Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cellular and Molecular Life Sciences, vol. 75, n°18, p. 3313‐3327.
- Smith, M. H., Ploegh, H. L. et Weissman, J. S. (2011) Road to Ruin: Targeting Proteins for Degradation in the Endoplasmic Reticulum. Science, vol. 334, n°6059, p. 1086‐1090.
- Koo, S.-H. (2013) Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clinical and molecular hepatology, vol. 19, n°3, p. 210‐215.
- Coburn, C. T., Hajri, T., Ibrahimi, A. et Abumrad, N. A. (2001) Role of CD36 in Membrane Transport and Utilization of Long-Chain Fatty Acids by Different Tissues. Journal of Molecular Neuroscience, vol. 16, n°2‐3, p. 117‐122.
- Stahl, A. (2004) A current review of fatty acid transport proteins (SLC27). Pflügers Archiv, vol. 447, n°5, p. 722‐727.
- Pandit (2012) Drug-Induced Hepatotoxicity: A Review. Journal of Applied Pharmaceutical Science.
- Fan, J. et de Lannoy, I. A. M. (2014) Pharmacokinetics. Biochemical Pharmacology, Special Issue: Pharmacology in 21st Century Biomedical Research, vol. 87, n°1, p. 93‐120.
- Roberts, D. J. et Hall, R. I. (2013) Drug absorption, distribution, metabolism and excretion considerations in critically ill adults. Expert Opinion on Drug Metabolism & Toxicology, vol. 9, n°9, p. 1067‐1084
- Croom, E. (2012). Metabolism of Xenobiotics of Human Environments. Progress in Molecular Biology and Translational Science (Vol. 112, p. 31‐88). Elsevier.
- Döring, B. et Petzinger, E. (2014) Phase 0 and phase III transport in various organs: Combined concept of phases in xenobiotic transport and metabolism. Drug Metabolism Reviews, vol. 46, n°3, p. 261‐282.
- Evans, W. E. et Relling, M. V. (1999) Pharmacogenomics: Translating Functional Genomics into Rational Therapeutics. Science, vol. 286, n°5439, p. 487‐491.
- Roth, A. D. et Lee, M.-Y. (2017) Idiosyncratic Drug-Induced Liver Injury (IDILI): Potential Mechanisms and Predictive Assays. BioMed Research International, vol. 2017.
- Jancova, P., Anzenbacher, P. et Anzenbacherova, E. (2010) PHASE II DRUG METABOLIZING ENZYMES. Biomedical Papers, vol. 154, n°2, p. 103‐116.
- Yasuhisa, K., Shin‐ya, M., Michinori, M. et Kazumitsu, U. (2007) Mechanism of multidrug recognition by MDR1/ABCB1. Cancer Science, vol. 98, n°9, p. 1303‐1310.
- Kock, K. et Brouwer, K. L. R. (2012) A Perspective on Efflux Transport Proteins in the Liver. Clinical pharmacology and therapeutics, vol. 92, n°5, p. 599‐612.
- Schapira, A. H. (2006) Mitochondrial disease. The Lancet, vol. 368, n°9529, p. 70‐82.
- Carracedo, A., Cantley, L. C. et Pandolfi, P. P. (2013) Cancer metabolism: fatty acid oxidation in the limelight. Nature Reviews. Cancer, vol. 13, n°4, p. 227‐232.
- Hashimoto, T., Cook, W. S., Qi, C., Yeldandi, A. V., Reddy, J. K. et Rao, M. S. (2000) Defect in Peroxisome Proliferator-activated Receptor α-inducible Fatty Acid Oxidation Determines the Severity of Hepatic Steatosis in Response to Fasting. Journal of Biological Chemistry, vol. 275, n°37, p. 28918‐28928.
- Reddy, J. K. et Hashimoto, T. (2001) Peroxisomal β-oxidation and peroxisome proliferatore-acivated receptor α: an adaptive metabolic system. Annual Review of Nutrition, vol. 21, n°1, p. 193‐230.
- Nsiah-Sefaa, A. et McKenzie, M. (2016) Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Bioscience Reports, vol. 36, n°2.
- Houten, S. M., Violante, S., Ventura, F. V. et Wanders, R. J. A. (2016) The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annual Review of Physiology, vol. 78, n°1, p. 23‐44.
- Houten, S. M., Wanders, R. J. A. et Ranea-Robles, P. (2020) Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease, vol. 1866, n°5, p. 165720.
- Schönfeld, P. et Wojtczak, L. (2016) Short- and medium-chain fatty acids in energy metabolism: the cellular perspective. Journal of Lipid Research, vol. 57, n°6, p. 943‐954.
- Fromenty, B. (2019) Inhibition of mitochondrial fatty acid oxidation in drug-induced hepatic steatosis. Liver Research.
- Longo, N., Filippo, C. A. di S. et Pasquali, M. (2006) Disorders of carnitine transport and the carnitine cycle. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, vol. 142C, n°2, p. 77‐85.
- Wanders, R. J. A., Ruiter, J. P. N., IJlst, L., Waterham, H. R. et Houten, S. M. (2010) The enzymology of mitochondrial fatty acid beta-oxidation and its application to follow-up analysis of positive neonatal screening results. Journal of Inherited Metabolic Disease, vol. 33, n°5, p. 479‐494.
- Adeva-Andany, M. M., Carneiro-Freire, N., Seco-Filgueira, M., Fernández-Fernández, C. et Mouriño- Bayolo, D. (2019) Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion, vol. 46, p. 73‐90.
- Begriche, K., Massart, J., Robin, M.-A., Bonnet, F. et Fromenty, B. (2013) Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology (Baltimore, Md.), vol. 58, n°4, p. 1497‐1507.
- Kersten, S. et Stienstra, R. (2017) The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie, «Pleiotropic physiological roles of PPARs and Fatty Acids» a tribute to Paul Grimaldi’, vol. 136, p. 75‐84.
- Klaunig, J. E., Li, X. et Wang, Z. (2018) Role of xenobiotics in the induction and progression of fatty liver disease. Toxicology Research, vol. 7, n°4, p. 664‐680.
- Massart, J., Begriche, K., Buron, N., Porceddu, M., Borgne-Sanchez, A. et Fromenty, B. (2013) Drug-Induced Inhibition of Mitochondrial Fatty Acid Oxidation and Steatosis. Current Pathobiology Reports, vol. 1, n°3, p. 147‐157.
- Massart, J., Begriche, K., Moreau, C. et Fromenty, B. (2017) Role of nonalcoholic fatty liver disease as risk factor for drug-induced hepatotoxicity. Journal of Clinical and Translational Research, vol. 3, n°Suppl 1, p. 212‐232.
- McGarry, J. D. et Brown, N. F. (1997) The Mitochondrial Carnitine Palmitoyltransferase System — From Concept to Molecular Analysis. European Journal of Biochemistry, vol. 244, n°1, p. 1‐14.
- Drosatos, K. et Schulze, P. C. (2013) Cardiac Lipotoxicity: Molecular Pathways and Therapeutic Implications. Current Heart Failure Reports, vol. 10, n°2, p. 109‐121.
- Wallace, D. C., Fan, W. et Procaccio, V. (2010) Mitochondrial Energetics and Therapeutics. Annual review of pathology, vol. 5, p. 297‐348.
- Kanabus, M., Heales, S. J. et Rahman, S. (2014) Development of pharmacological strategies for mitochondrial disorders. British Journal of Pharmacology, vol. 171, n°8, p. 1798‐1817.
- Wang, Yuhui, Viscarra, J., Kim, S.-J. et Sul, H. S. (2015) Transcriptional regulation of hepatic lipogenesis. Nature Reviews Molecular Cell Biology, vol. 16, n°11, p. 678‐689.
- Yamashita, H., Takenoshita, M., Sakurai, M., Bruick, R. K., Henzel, W. J., Shillinglaw, W., Arnot, D. et Uyeda, K. (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proceedings of the National Academy of Sciences of the United States of America, vol. 98, n°16, p. 9116‐9121.
- Iizuka, K., Bruick, R. K., Liang, G., Horton, J. D. et Uyeda, K. (2004) Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proceedings of the National Academy of Sciences of the United States of America, vol. 101, n°19, p. 7281‐7286.
- Dentin, R., Tomas-Cobos, L., Foufelle, F., Leopold, J., Girard, J., Postic, C. et Ferré, P. (2012) Glucose 6- phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. Journal of Hepatology, vol. 56, n°1, p. 199‐209.
- Arden, C., Tudhope, S. J., Petrie, J. L., Al-Oanzi, Z. H., Cullen, K. S., Lange, A. J., Towle, H. C. et Agius, L. (2012) Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. The Biochemical Journal, vol. 443, n°1, p. 111‐123.
- Horton, J. D., Goldstein, J. L. et Brown, M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. The Journal of Clinical Investigation, vol. 109, n°9, p. 1125‐1131.
- Lin, J., Yang, R., Tarr, P. T., Wu, P.-H., Handschin, C., Li, S., Yang, W., Pei, L., Uldry, M., Tontonoz, P., Newgard, C. B. et Spiegelman, B. M. (2005) Hyperlipidemic Effects of Dietary Saturated Fats Mediated through PGC-1β Coactivation of SREBP. Cell, vol. 120, n°2, p. 261‐273.
- Caron, S., Huaman Samanez, C., Dehondt, H., Ploton, M., Briand, O., Lien, F., Dorchies, E., Dumont, J., Postic, C., Cariou, B., Lefebvre, P. et Staels, B. (2013) Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Molecular and Cellular Biology, vol. 33, n°11, p. 2202‐2211.
- Cha, J.-Y. et Repa, J. J. (2007) The Liver X Receptor (LXR) and Hepatic Lipogenesis THE CARBOHYDRATE- RESPONSE ELEMENT-BINDING PROTEIN IS A TARGET GENE OF LXR. Journal of Biological Chemistry, vol. 282, n°1, p. 743‐751.
- Watanabe, M., Houten, S. M., Wang, L., Moschetta, A., Mangelsdorf, D. J., Heyman, R. A., Moore, D. D. et Auwerx, J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. Journal of Clinical Investigation, vol. 113, n°10, p. 1408‐1418.
- Liu, S., Hatano, B., Zhao, M., Yen, C.-C., Kang, K., Reilly, S. M., Gangl, M. R., Gorgun, C., Balschi, J. A., Ntambi, J. M. et Lee, C.-H. (2011) Role of Peroxisome Proliferator-activated Receptor δ/β in Hepatic Metabolic Regulation. The Journal of Biological Chemistry, vol. 286, n°2, p. 1237‐1247.
- Lee, Y. J., Ko, E. H., Kim, J. E., Kim, E., Lee, H., Choi, H., Yu, J. H., Kim, H. J., Seong, J.-K., Kim, K.-S. et Kim, J. (2012) Nuclear receptor PPARγ-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proceedings of the National Academy of Sciences of the United States of America, vol. 109, n°34, p. 13656‐13661.
- Sanders, F. W. B. et Griffin, J. L. (2016) De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biological Reviews of the Cambridge Philosophical Society, vol. 91, n°2, p. 452‐468.
- Gluchowski, N. L., Becuwe, M., Walther, T. C. et Farese, R. V. (2017) Lipid droplets and liver disease: from basic biology to clinical implications. Nature Reviews Gastroenterology & Hepatology, vol. 14, n°6, p. 343‐355.
- Takeuchi, K. et Reue, K. (2009) Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. American Journal of Physiology – Endocrinology and Metabolism, vol. 296, n°6, p. E1195‐E1209.
- Brasaemle, D. L. (2007) Thematic review series: Adipocyte Biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. Journal of Lipid Research, vol. 48, n°12, p. 2547‐2559.
- Choudhary, V., Ojha, N., Golden, A. et Prinz, W. A. (2015) A conserved family of proteins facilitates nascent lipid droplet budding from the ER. The Journal of Cell Biology, vol. 211, n°2, p. 261‐271.
- Wilfling, F., Wang, H., Haas, J. T., Krahmer, N., Gould, T. J., Uchida, A., Cheng, J.-X., Graham, M., Christiano, R., Fröhlich, F., Liu, X., Buhman, K. K., Coleman, R. A., Bewersdorf, J., Farese, R. V. et Walther, T. C. (2013) Triacylglycerol Synthesis Enzymes Mediate Lipid Droplet Growth by Relocalizing from the ER to Lipid Droplets. Developmental cell, vol. 24, n°4, p. 384‐399.
- Gordon, D. A., Wetterau, J. R. et Gregg, R. E. (1995) Microsomal triglyceride transfer protein: a protein complex required for the assembly of lipoprotein particles. Trends in Cell Biology, vol. 5, n°8, p. 317‐321.
- Balaban, S., Lee, L. S., Schreuder, M. et Hoy, A. J. (2015) Obesity and Cancer Progression: Is There a Role of Fatty Acid Metabolism? BioMed Research International. Research article.
- Buqué, X., Cano, A., Miquilena-Colina, M. E., García-Monzón, C., Ochoa, B. et Aspichueta, P. (2012) High insulin levels are required for FAT/CD36 plasma membrane translocation and enhanced fatty acid uptake in obese Zucker rat hepatocytes. American Journal of Physiology – Endocrinology and Metabolism, vol. 303, n°4, p. E504‐E514.
- Glatz, J. F. C. et Luiken, J. J. F. P. (2018) Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. Journal of Lipid Research, vol. 59, n°7, p. 1084‐1093.
- Wong, H. et Schotz, M. C. (2002) The lipase gene family. Journal of Lipid Research, vol. 43, n°7, p. 993‐999.
- Højmann Larsen, A., Frandsen, A. et Treiman, M. (2001) Upregulation of the SERCA-type Ca2+ pump activity in response to endoplasmic reticulum stress in PC12 cells. BMC Biochemistry, vol. 2, p. 4.
- Chen, S., Melchior, W. B. et Guo, L. (2014) Endoplasmic Reticulum Stress in Drug- and Environmental Toxicant-Induced Liver Toxicity. Journal of environmental science and health. Part C, Environmental carcinogenesis & ecotoxicology reviews, vol. 32, n°1, p. 83‐104.
- Dara, L., Ji, C. et Kaplowitz, N. (2011) The contribution of endoplasmic reticulum stress to liver diseases. Hepatology (Baltimore, Md.), vol. 53, n°5, p. 1752‐1763.
- Foufelle, F. et Fromenty, B. (2016) Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacology Research & Perspectives, vol. 4, n°1, p. e00211.
- Kabir, M. F., Kim, H.-R. et Chae, H.-J. (2018) Endoplasmic Reticulum Stress and Autophagy. Endoplasmic Reticulum.
- Liu, Z., Lv, Y., Zhao, N., Guan, G. et Wang, J. (2015) Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death & Disease, vol. 6, n°7, p. e1822‐e1822.
- Urano, F., Wang, X., Bertolotti, A., Zhang, Y., Chung, P., Harding, H. P. et Ron, D. (2000) Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science, vol. 287, n°5453, p. 664‐666.
- Wang, S. et Kaufman, R. J. (2014) How does protein misfolding in the endoplasmic reticulum affect lipid metabolism in the liver?: Current Opinion in Lipidology, vol. 25, n°2, p. 125‐132.
- Wang, Yan, Gusarova, V., Banfi, S., Gromada, J., Cohen, J. C. et Hobbs, H. H. (2015) Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion 1. Journal of Lipid Research, vol. 56, n°7, p. 1296‐1307.
- Hollien, J. et Weissman, J. S. (2006) Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science, vol. 313, n°5783, p. 104‐107.
- Oakes, S. A. et Papa, F. R. (2015) The Role of Endoplasmic Reticulum Stress in Human Pathology. Annual Review of Pathology: Mechanisms of Disease, vol. 10, n°1, p. 173‐194.
- Ron, D. et Walter, P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology, vol. 8, n°7, p. 519‐529.
- Wang, S. et Kaufman, R. J. (2012) The impact of the unfolded protein response on human disease. The Journal of Cell Biology, vol. 197, n°7, p. 857‐867.
- Oslowski, C. M. et Urano, F. (2010) The binary switch between life and death of endoplasmic reticulum- stressed β cells: Current Opinion in Endocrinology, Diabetes and Obesity, vol. 17, n°2, p. 107‐112.
- Pobre, K. F. R., Poet, G. J. et Hendershot, L. M. (2018) The endoplasmic reticulum (ER) chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. Journal of Biological Chemistry, p. jbc.REV118.002804.
- Yoshida, H., Matsui, T., Yamamoto, A., Okada, T. et Mori, K. (2001) XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell, vol. 107, n°7, p. 881‐891.
- Lee, A.-H., Scapa, E. F., Cohen, D. E. et Glimcher, L. H. (2008) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science (New York, N.Y.), vol. 320, n°5882, p. 1492‐1496.
- Anelli, T. et Sitia, R. (2008) Protein quality control in the early secretory pathway. The EMBO Journal, vol. 27, n°2, p. 315‐327.
- Rutledge, A. C., Su, Q. et Adeli, K. (2010) Apolipoprotein B100 biogenesis: a complex array of intracellular mechanisms regulating folding, stability, and lipoprotein assembly. Cet article fait partie d’une sélection d’articles publiés dans ce numéro spécial intitulé “Canadian Society of Biochemistry, Molecular & Cellular Biology 52nd Annual Meeting — Protein Folding: Principles and Diseases” et a été soumis au processus habituel d’évaluation par les pairs du Journal. Biochemistry and Cell Biology, vol. 88, n°2, p. 251‐267.
- Jiao, Y., Lu, Y. et Li, X. (2015) Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharmacologica Sinica, vol. 36, n°1, p. 44‐50.
- Lee, G., Jeong, Y. S., Kim, D. W., Kwak, M. J., Koh, J., Joo, E. W., Lee, J.-S., Kah, S., Sim, Y.-E. et Yim, S. Y. (2018) Clinical significance of APOB inactivation in hepatocellular carcinoma. Experimental & Molecular Medicine, vol. 50, n°11, p. 1‐12.
- Jiang, Z. G., Robson, S. C. et Yao, Z. (2013) Lipoprotein metabolism in nonalcoholic fatty liver disease. Journal of Biomedical Research, vol. 27, n°1, p. 1‐13.
- Fisher E, Lake E, McLeod RS. Apolipoprotein B100 quality control and the regulation of hepatic very low density lipoprotein secretion. J Biomed Res. 2014;28(3):178-93.
- Cefalù, A. B., Pirruccello, J. P., Noto, D., Gabriel, S., Valenti, V., Gupta, N., Spina, R., Tarugi, P., Kathiresan, S. et Averna, M. R. (2013) A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer and hypocholesterolemia. Arteriosclerosis, thrombosis, and vascular biology, vol. 33, n°8.
- Welty, F. K. (2014) Hypobetalipoproteinemia and Abetalipoproteinemia. Current opinion in lipidology, vol. 25, n°3, p. 161‐168.
- Hossain, T., Riad, A., Siddiqi, S., Parthasarathy, S. et Siddiqi, S. A. (2014) Mature VLDL triggers the biogenesis of a distinct vesicle from the trans-Golgi network for its export to the plasma membrane. The Biochemical journal, vol. 459, n°1, p. 47‐58.
- Tiwari, S. et Siddiqi, S. A. (2012) Intracellular Trafficking and Secretion of Very Low Density Lipoproteins. Arteriosclerosis, Thrombosis, and Vascular Biology, vol. 32, n°5, p. 1079‐1086.
- Tiwari, S., Siddiqi, S. et Siddiqi, S. A. (2013) CideB protein is required for the biogenesis of very low density lipoprotein (VLDL) transport vesicle. The Journal of biological chemistry, vol. 288, n°7, p. 5157‐5165.
- Yao, Z. et Wang, Y. (2012) Apolipoprotein C-III and hepatic triglyceride-rich lipoprotein production: Current Opinion in Lipidology, vol. 23, n°3, p. 206‐212.
- Olofsson, S.-O., Bjursell, G., Boström, K., Carlsson, P., Elovson, J., Protter, A. A., Reuben, M. A. et Bondjers, G. (1987) Apolipoprotein B: structure, biosynthesis and role in the lipoprotein assembly process. Atherosclerosis, vol. 68, n°1, p. 1‐17.
- Olofsson, S.-O., Stillemark-Billton, P. et Asp, L. (2000) Intracellular Assembly of VLDL: Two Major Steps in Separate Cell Compartments. Trends in Cardiovascular Medicine, vol. 10, n°8, p. 338‐345.
- Elovson, J., Chatterton, J. E., Bell, G. T., Schumaker, V. N., Reuben, M. A., Puppione, D. L., Reeve, J. R. et Young, N. L. (1988) Plasma very low density lipoproteins contain a single molecule of apolipoprotein B. Journal of Lipid Research, vol. 29, n°11, p. 1461‐1473.
- Mobin, M. B., Gerstberger, S., Teupser, D., Campana, B., Charisse, K., Heim, M. H., Manoharan, M., Tuschl, T. et Stoffel, M. (2016) The RNA-binding protein vigilin regulates VLDL secretion through modulation of Apob mRNA translation. Nature Communications, vol. 7, n°1, p. 1‐13.
- Alves-Bezerra, M. et Cohen, D. E. (2017) Triglyceride metabolism in the liver. Comprehensive Physiology, vol. 8, n°1, p. 1‐8.
- Trefts E, Gannon M, Wasserman DH. The liver. Curr Biol. 2017 Nov 6;27(21):R1147-R1151. doi: 10.1016/j.cub.2017.09.019. PMID: 29112863; PMCID: PMC5897118.
- SNFGE. Foie – Voies biliaires. In: Les fondamentaux de la pathologie digestive [Internet]. Elsevier Masson. 2014. p. 39.
- Hossain, T., Riad, A., Siddiqi, S., Parthasarathy, S. et Siddiqi, S. A. (2014) Mature VLDL triggers the biogenesis of a distinct vesicle from the trans-Golgi network for its export to the plasma membrane. The Biochemical journal, vol. 459, n°1, p. 47‐58.
- Mauss, Berg, Rockstroh, Sarrazin, Wedemeyer, Hepatology. A clinical textbook. 10th Edition 2020.