/édition génique de l'endonucléase associée à CRISPR (Cas9) [méganucléases, doigts de zinc et nucléases effectrices de type activateur de transcription (TALEN)] ; le développement de CRISPR/Cas9 ; la précision de l’édition génétique ; l’amélioration de la spécificité de CRISPR/Cas9; le contrôle qualité et assurance qualité pour l’édition génétique ; l’utilisation de Cas9 morts (dCas9) pour réguler la transcription ou pour apporter des modifications épigénétiques ; le ciblage génique chez les animaux transgéniques ; l’édition génétique chez les embryons ; les voies alternatives à l’édition de la lignée germinale héréditaire; ainsi que sur la modification du génome mitochondrial.){kind=link}
Cet article porte su la science fondamentale de la thérapie génique et de l’édition génétique notamment sur les dommages et réparation de l’ADN ; les précurseurs du système de répétitions palindromiques courtes et régulièrement intercalées en cluster (CRISPR)/édition génique de l’endonucléase associée à CRISPR (Cas9) [méganucléases, doigts de zinc et nucléases effectrices de type activateur de transcription (TALEN)] ; le développement de CRISPR/Cas9 ; la précision de l’édition génétique ; l’amélioration de la spécificité de CRISPR/Cas9; le contrôle qualité et assurance qualité pour l’édition génétique ; l’utilisation de Cas9 morts (dCas9) pour réguler la transcription ou pour apporter des modifications épigénétiques ; le ciblage génique chez les animaux transgéniques ; l’édition génétique chez les embryons ; les voies alternatives à l’édition de la lignée germinale héréditaire; ainsi que sur la modification du génome mitochondrial.
Source : National Academies of Sciences, Engineering, and Medicine. 2017. Human Genome Editing: Science, Ethics, and Governance. Washington, DC: The National Academies Press. https://doi.org/10.17226/24623.
1. Thérapie génique et édition du génome
Le potentiel de la thérapie génique pour lutter contre les maladies humaines est évident depuis quelques années, et de nombreux progrès ont été réalisés dans ses applications (Cox et al., 2015 ; Naldini, 2015). La thérapie génique fait référence au remplacement de gènes défectueux ou à l’ajout de nouveaux gènes comme moyen de guérir une maladie ou d’améliorer la capacité à combattre la maladie.
L’édition du génome est un aspect de la thérapie génique. Les approches établies en matière de thérapie génique reposent sur les résultats de recherches approfondies en laboratoire sur des cellules individuelles et sur des organismes non humains, établissant les moyens d’ajouter, de supprimer ou de modifier des gènes dans les organismes vivants. Les principales avancées comprennent le développement de techniques permettant de générer des outils moléculaires permettant de couper l’ADN des génomes à des endroits spécifiques afin de permettre des modifications ciblées de la séquence d’ADN. Au cours des dernières années, plusieurs de ces méthodes ont été introduites et utilisées efficacement dans des applications cliniques.
Au cours des cinq dernières années, un système totalement nouveau a été développé sur la base de la recherche fondamentale sur les systèmes bactériens d’immunité contre les infections virales. Le premier système de ce type développé pour être utilisé dans l’édition du génome de cellules humaines, connu sous le nom de CRISPR/Cas9, est basé sur le ciblage guidé par l’ARN et est beaucoup plus simple, plus rapide et moins cher que les méthodes précédentes. La facilité de conception, ainsi que la spécificité et l’efficacité remarquables du système CRISPR/Cas9 ont révolutionné le domaine de l’édition du génome et relancé l’intérêt pour le potentiel d’édition du génome humain. Le développement du système CRISPR/Cas9 en tant qu’outil programmable d’édition du génome repose sur une base solide de recherches antérieures.
2. Dommages et réparation de l’ADN génomique
Les génomes et leurs gènes constitutifs sont constitués d’ADN double brin ; cet ADN peut être brisé accidentellement (par exemple par rayonnement) ou volontairement, à l’aide de protéines appelées endonucléases (souvent appelées nucléases) qui peuvent générer des cassures double brin (DSB) dans l’ADN.
Les cellules disposent de mécanismes pour réparer les DSB dans l’ADN, et ces mécanismes peuvent être utilisés pour générer des altérations dans la séquence d’ADN. Des travaux révolutionnaires sur les systèmes de bactéries, de levures et de mammifères montrent que les DSB stimulent considérablement le taux de réparation de l’ADN par assemblage d’extrémités non homologues (NHEJ), dans lequel les extrémités cassées sont rattachées (figure 1). Une telle réparation du NHEJ entraîne souvent la suppression ou l’insertion de séquences d’ADN de longueur variable, ce qui peut perturber la fonction des gènes (Rouet et al., 1994).
Cependant, si une séquence homologue d’ADN est introduite dans la cellule en tant que modèle donneur, la réparation dirigée par homologie (HDR) peut conduire à une réparation plus précise ou, si des altérations spécifiques sont incluses dans l’étirement homologue, cela peut introduire des changements spécifiques et précis dans l’ADN génomique du receveur (figure 1). Ces mécanismes de réparation de l’ADN cellulaire ont été utilisés pour développer plusieurs méthodes permettant d’éditer les gènes ou le génome de manière très précise.
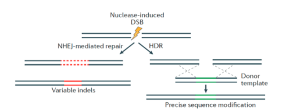
Figure 1: Les résultats de l’édition du génome sont médiés par la réparation des cassures d’ADN double brin induites par des nucléases par NHEJ ou HDR. DSB = rupture double brin ; HDR = réparation dirigée par homologie; NHEJ = jointure d’extrémité non homologue. © Modifié à partir de Sander et Joung, 2014.
3. Précurseurs de l’édition du gène par CRISPR/CAS9
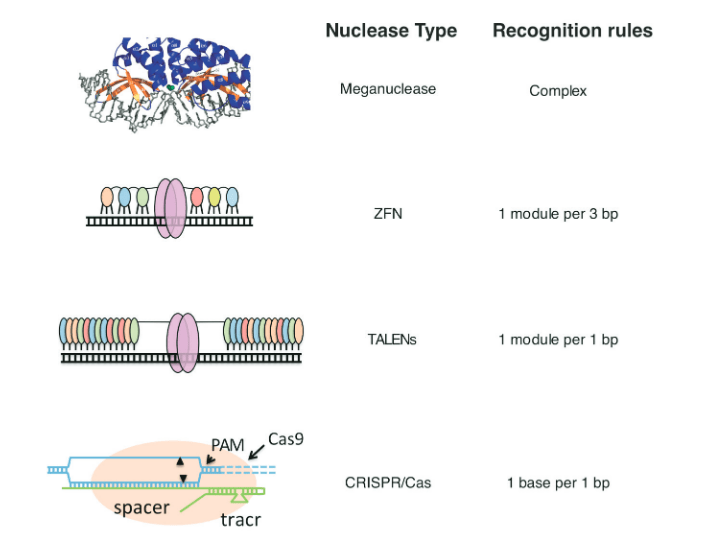
Trois stratégies distinctes basées sur des systèmes de nucléases pour générer des clivages ciblés dans l’ADN ont précédé le développement de CRISPR/Cas9 : les méganucléases, les nucléases à doigts de zinc (ZFN) et les TALEN (figure 2). Tous trois ont déjà permis des avancées majeures dans l’établissement de la faisabilité de l’utilisation de telles nucléases ciblées à la fois pour éliminer les gènes pathogènes et pour réparer les gènes endommagés ou mutés, ouvrant ainsi la voie à une nouvelle ère en biologie et en médecine.
3.1. Nucléases à doigts de zinc
Les doigts de zinc sont des segments de protéines qui ont évolué pour reconnaître et se lier à des séquences d’ADN spécifiques. Les connaissances acquises grâce aux doigts de zinc naturels ont conduit au développement des ZFN en tant qu’enzymes de conception coupant l’ADN (figure 2), et leur utilisation dans l’ingénierie du génome représente une ingénierie pionnière, voire héroïque, des protéines. Deux avancées majeures dans l’ingénierie des protéines ont permis le développement des ZFN : (1) l’ingénierie des protéines à doigt de zinc avec une spécificité de liaison à l’ADN conçue par Berg (Desjarlais et Berg, 1992), Pabo (Rebar et Pabo, 1994) et Wells (Jamieson et al., 1994); et (2) la génération de fusions entre ces doigts de zinc de conception et une protéine de clivage de l’ADN, la nucléase FokI (Kim et al., 1996), produisant des «enzymes de restriction artificielles» qui pourraient être utilisées pour promouvoir l’ingénierie génomique spécifique à un site en créant DSB à des emplacements définis dans le génome (Bibikova et al., 2001, 2002, 2003).
Grâce à ces résultats, Sangamo Therapeutics a développé des ZFN à partir d’outils de laboratoire pour en faire des agents thérapeutiques qui pourraient être utilisés pour désactiver les gènes responsables de maladies (par NHEJ) ou pour corriger des erreurs dans les gènes existants (par HDR). Ce dernier objectif est particulièrement difficile, car l’efficacité du HDR est généralement bien inférieure à la réparation imprécise et mutagène du NHEJ. En surmontant certains de ces défis difficiles, Sangamo a désormais plusieurs essais cliniques en cours sur l’homme, dont le plus avancé concerne le traitement du VIH/SIDA, où la suppression du corécepteur CCR5 du VIH a le potentiel de permettre l’élimination du VIH après une transplantation de la moelle osseuse. D’autres applications des ZFN ont également été ou sont en cours de test dans des essais cliniques (NCT02695160 [hémophilie B] et NCT02702115 [édition in vivo MPS1]).
3.2. Nucléases effectrices de type activateur de transcription
Comme les ZFN, les TALEN sont composés d’une protéine de liaison à l’ADN qui reconnaît des séquences d’ADN particulières fusionnées à un domaine effecteur de nucléase pour réaliser le clivage (Joung et Sander, 2013) (figure 2). Les TALE sont des protéines bactériennes sécrétées avec des domaines de liaison à l’ADN qui contiennent une série de blocs conservés de séquence de 32 à 34 résidus de long, chacun avec deux acides aminés divergents. Ces acides aminés divergents sont en grande partie responsables de la détermination de la spécificité de liaison à l’ADN pour une seule paire de bases d’ADN, ce qui permet l’ingénierie de domaines spécifiques de liaison à l’ADN en choisissant des combinaisons de segments répétés contenant les acides aminés appropriés. La société de biotechnologie Cellectis a signalé la conduite réussie du tout premier essai clinique d’édition de gènes basé sur TALEN au Royaume-Uni sur une fille atteinte de leucémie lymphoblastique aiguë (LAL) incurable (Qasim et al, 2017). Bien que les applications des ZFN et des TALEN se chevauchent largement, les TALENS présentent l’avantage d’une relative facilité de conception grâce à leur code de reconnaissance robuste.
3.3. Méganucléases
Les méganucléases sont des nucléases dotées de très longs sites de reconnaissance de liaison à l’ADN, jusqu’à 40 nucléotides (Silva et al., 2011) (figure 2). En raison de leur longueur, il est extrêmement improbable que des sites naturels soient présents par hasard, même dans des génomes humains complexes. Le défi des méganucléases réside dans la difficulté de concevoir de nouvelles nucléases pour cibler à volonté une séquence d’intérêt. Un certain succès a été obtenu grâce à des modifications conçues des sites de liaison à l’ADN et à la combinaison de méganucléases avec des éléments de liaison à l’ADN TALE. Cependant, il est peu probable que les méganucléases soient très utilisées dans l’édition du génome humain étant donné la relative simplicité des méthodes alternatives.
4. Développement de CRISPR/CAS9
4.1. CRISPR comme système d’immunité adaptative bactérienne
La découverte que les systèmes CRISPR confèrent une immunité adaptative aux bactéries représente en soi une avancée conceptuelle majeure. Cette découverte a également été essentielle au développement de l’ingénierie du génome CRISPR/Cas9. Un bref résumé des principales conclusions est présenté ci-dessous (pour un examen plus complet, voir Doudna et Charpentier, 2014).
Les loci CRISPR ont été identifiés pour la première fois sur la base d’analyses de génomes bactériens. De ces études, il a été déduit que les régions espaceurs (c’est-à-dire non répétitives) des loci CRISPR étaient dérivées de l’ADN génomique des bactériophages (virus qui infectent les bactéries), conduisant à l’hypothèse que CRISPR fournissait un mécanisme de défense contre les éléments génétiques étrangers ( Makarova et al., 2006 ; Mojica et al., 2005 ; Pourcel et al., 2005).
La principale percée expérimentale est venue de recherches montrant que CRISPR permettait aux bactéries d’acquérir une résistance aux bactériophages en intégrant des segments du génome du bactériophage dans les locus CRISPR, démontrant que CRISPR était une nouvelle forme d’immunité adaptative (Barrangou et al., 2007). En 2010, il a été démontré que les systèmes CRISPR/Cas de type II intervenaient dans le clivage de l’ADN des bactériophages envahisseurs (Garneau et al., 2010). Et en 2011, un ARN associé, tracrRNA, a été identifié (Deltcheva et al., 2011), et le gène associé à CRISPR, Cas9, s’est avéré être le seul gène codant pour une protéine dans le locus Cas de type II requis pour la défense. fonction (Sapranauskas et al., 2011).
4.2. Développement de Cas9 en tant qu’endonucléase programmable
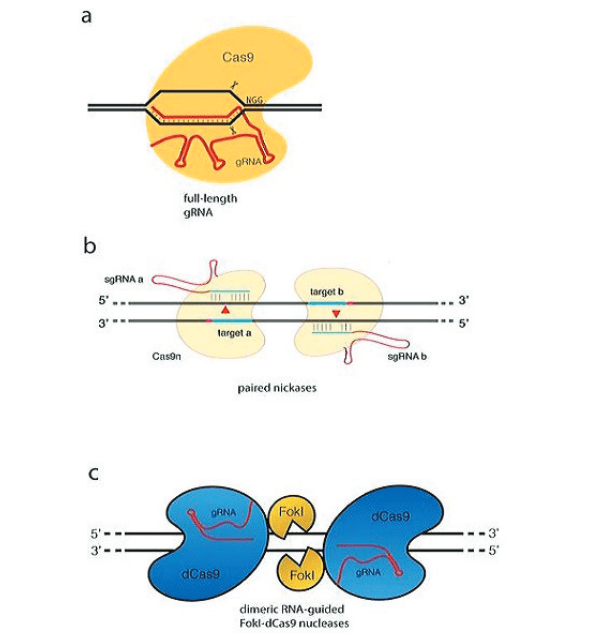
L’avancée cruciale dans le développement de la méthode d’édition génomique basée sur CRISPR/Cas9 est venue en 2012 des laboratoires de Doudna et Charpentier. Ils ont établi que la protéine Cas9 associée à CRISPR, en complexe avec deux petits ARN, l’ARN CRISPR (ARNcr) transcrit à partir du locus CRISPR et un ARNcr trans-activant (tracrARN), produit une endonucléase spécifique au site dans laquelle le site de clivage est défini par l’appariement des bases de l’ARNcr à l’ADN cible. Cela a jeté les bases pour établir que Cas9 est une endonucléase qui peut être programmée avec un seul «ARN guide» (ARNg, une chimère d’ARNcr transcrit à partir du locus CRISPR et du tracrRNA) pour se diviser sur des sites d’ADN spécifiques (Jinek et al., 2012) (figure 3a).
Concurrent avec Jinek et al. (2012), Siksnys et ses collègues (Gasiunas et al., 2012) ont démontré que des complexes purifiés contenant Cas9 et un ARNc pouvaient médier le clivage de l’ADN double brin in vitro sur des sites complémentaires de l’ARNc. Bien qu’il s’agisse clairement d’un système Cas9, il est supérieur aux ZFN et aux TALEN en termes de facilité d’utilisation ; tout ce qui est nécessaire pour générer une nucléase spécifique à un site est la conception et la synthèse d’un seul ARNg pour cibler la nucléase Cas9 vers le site d’édition souhaité. Bien que tous les ARN guides prédits ne fonctionnent pas, leur synthèse est facile et plusieurs candidats possibles peuvent être synthétisés facilement et à moindre coût.
4.3. Application in vivo de la nucléase programmable Cas9
Quelques mois après la publication de Jinek et al. (2012) et dans les manuscrits du laboratoire Siksnys (Gasiunas et al., 2012; Sapranauskas et al., 2011), six rapports indépendants ont été publiés utilisant le système d’ARN guide Cas9 pour assurer l’édition programmable du génome in vivo. Ceux-ci comprenaient quatre articles rapportant l’édition de Cas9 dans des cellules de mammifères (Cho et al., 2013 ; Cong et al., 2013 ; Jinek et al., 2013 ; Mali et al., 2013) ; un sur le poisson zèbre (Hwang et al., 2013) ; et un sur les bactéries (Jiang et al., 2013). De plus, un septième article (Qi et al., 2013) montre que dCas9 pourrait être utilisé pour inhiber la transcription. Depuis lors, il y a eu une explosion dans l’application et le perfectionnement du clivage médié par Cas9, ainsi que la découverte de nouveaux systèmes CRISPR pouvant être adaptés pour l’édition du génome. Il s’agit notamment de la découverte d’une nouvelle endonucléase guidée par l’ARN, Cpf1 (Zetsche et al., 2015), et des travaux plus récents ont montré que d’autres nucléases ciblables par CRISPR et présentant un potentiel de capacités différentes sont en cours de découverte.
5. Précision de l’édition des gène
L’impact potentiel des modifications involontaires de l’ADN constitue un défi majeur pour une utilisation sûre de l’édition du génome comme stratégie thérapeutique. Des modifications involontaires du génome pourraient être provoquées par le clivage de l’ADN sur des sites autres que ceux délibérément ciblés.
5.1. Le défi de la toxicité hors cible
Le site sur lequel Cas9 clive l’ADN est déterminé par la complémentarité de l’ADN cible avec le guide d’ARN (généralement 20 paires de bases) adjacent à une séquence de motif adjacent au protoespaceur (PAM) (par exemple, NGG pour Streptococcus pyogenes, le plus couramment utilisé). espèce de Cas9). En principe, cette séquence de 22 bases donnerait suffisamment de diversité pour que le site de coupure soit unique, même au sein d’un génome humain de 3 milliards de paires de bases. Dans la pratique, cependant, certaines disparités de base sont tolérées, ce qui entraîne un risque important de coupure hors cible. Cela a motivé les efforts à la fois pour surveiller les sites de coupure hors cible et pour améliorer la spécificité des nucléases ciblées. Les efforts initiaux visant à définir la coupe hors cible de Cas9 se sont concentrés sur des recherches spécifiques de coupe sur des sites proches. Plus récemment, ces efforts ont été complétés par des efforts moins biaisés à l’échelle du génome. Ceux-ci peuvent être divisés en deux grandes classes : à base de cellules et sans cellules (in vitro).
5.2. Essais cellulaires à l’échelle du génome
Apparemment, le séquençage du génome entier (WGS), lorsqu’il est effectué au niveau d’une seule cellule, semblerait fournir une évaluation définitive de la précision de l’édition du génome Cas9. Cependant, la profondeur du séquençage qui serait nécessaire pour certifier l’absence de coupure hors cible est actuellement difficile à atteindre pour les populations de cellules. Il devrait cependant être possible d’estimer la sensibilité du système pour détecter les modifications hors cible. L’incapacité de détecter l’édition avec le test indiquerait alors que le taux d’édition hors cible était inférieur au niveau de détection.
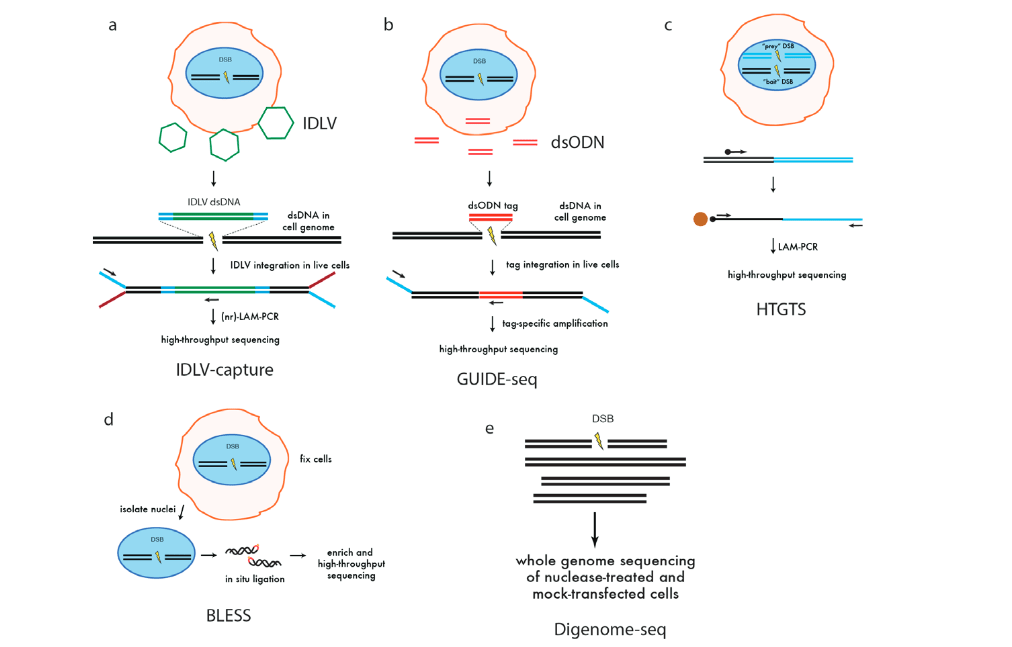
La capture de vecteurs lentiviraux déficients en intégrase (IDLV) (figure 4a) est une approche pangénomique utilisée pour évaluer la spécificité des nucléases d’édition du génome qui a été initialement appliquée aux ZFN modifiés, puis appliquée plus tard aux TALEN et CRISPR/Cas9 ( Gabriel et coll., 2011 ; Wang et coll., 2015). Cette méthode est basée sur la capture par NHEJ d’IDLV, qui possèdent des génomes d’ADN double brin linéaire, dans des sites de DSB induits par des nucléases. Bien que la méthode de capture IDLV identifie directement les DSB présents dans les cellules vivantes, elle est relativement insensible et possède un bruit de fond élevé. Pour surmonter ces limitations, une identification impartiale des DSB à l’échelle du génome, rendue possible par le séquençage (GUIDE-seq) (figure 4b), a été développée (Tsai et al., 2015). GUIDE-seq exploite l’intégration efficace d’une étiquette oligodésoxynucléotide double brin (dsODN) à extrémités émoussées et protégées, suivie d’une amplification spécifique de l’étiquette et d’un séquençage à haut débit. GUIDE-seq peut détecter les sites hors cible mutagénisés par les Cas9-sgRNA à basse fréquence (<0,1%) dans une population cellulaire, même avec seulement quelques millions de lectures de séquençage.
Le séquençage de translocation à haut débit à l’échelle du génome (HTGTS) (figure 4c) est une autre méthode à l’échelle du génome qui identifie le clivage hors cible de Cas9 dans les cellules vivantes (Chiarle et al., 2011). HTGTS est basé sur la détection de translocations entre un DSB «appât» induit par une nucléase et des DSB «proies» hors cible. Une limitation de HTGTS est que les translocations induites par des nucléases représentent des événements rares et nécessitent donc un grand nombre de génomes d’entrée pour la détection. Une stratégie de détection des DSB induits par des nucléases à l’échelle du génome dans des cellules fixes, appelée «BLESS» pour le marquage des ruptures, l’enrichissement en streptavidine et le séquençage de nouvelle génération, capture un instantané des DSB transitoires qui sont présents à un moment donné dans une population de cellules par ligature directe in situ d’adaptateurs en épingle à cheveux biotinylés dans des noyaux cellulaires fixes et perméabilisés (figure 4d).
5.3. Essais in vitro à l’échelle du génome
Digenome-seq (figure 4e) est une méthode in vitro de détection des DSB induits par des nucléases dans l’ADN génomique à l’aide du séquençage du génome entier de l’ADN génomique clivé par Cas9. L’ADN génomique est isolé et traité avec des concentrations élevées de Cas9 – ARNg in vitro pour maximiser le clivage hors cible et les sites de clivage sont identifiés par séquençage de l’ADN. Étant donné que ce test est effectué in vitro sur de l’ADN purifié, il n’est pas limité par des facteurs cellulaires tels que le contexte chromatinien, les facteurs épigénétiques, la localisation sous-nucléaire ou les effets de fitness. Digenome-seq peut ainsi détecter un clivage supplémentaire potentiel hors cible sur des sites qui autrement seraient masqués par les méthodes cellulaires. Ainsi, cela pourrait donner lieu à une surestimation des événements hors cible in vivo.
6. Amélioration de la spécificité de CRISPR/Cas9
Compte tenu de sa facilité d’utilisation, de sa flexibilité et de sa polyvalence, le système CRISPR/Cas9 devient rapidement l’outil de choix pour l’édition génétique. Cependant, les inquiétudes concernant le risque potentiel d’effets indésirables hors cible ont dominé de nombreuses discussions récentes. La plupart des expériences qui ont détecté des hors-cibles importants ont été réalisées dans des cellules cancéreuses (Fu et al., 2013 ; Hsu et al., 2013), ce qui peut avoir modifié les voies de réparation de l’ADN, ce qui pourrait conduire à une augmentation des événements hors cible. En revanche, des expériences sur des organismes entiers tels que des souris (Yang et al., 2013), des primates (Niu et al., 2014), des poissons zèbres (Auer et al., 2014) ou Caenorhabditis elegans (Dickinson et al., 2013) ont signalé des fréquences hors cible faibles ou non détectables, ce qui concorde avec la spécificité élevée du ciblage génique médié par CRISPR/Cas9. Il est possible que, dans les cellules non transformées, les clivages hors cible soient efficacement contre-sélectionnés par la réponse endogène aux dommages à l’ADN.
Les cellules souches pluripotentes humaines (hPSC) sont des cellules primaires dotées de mécanismes de contrôle de qualité génétiquement intacts, et il semble possible que les événements non ciblés s’accumulent moins fréquemment dans les hPSC ou dans les cellules somatiques normales que ce qui a été observé dans les cellules cancéreuses. Néanmoins, il sera important de déterminer s’il existe des types de cellules et des conditions spécifiques qui prédisposent à l’accumulation d’événements hors cible. Pour répondre aux préoccupations concernant les événements hors cible, diverses approches visant à minimiser les erreurs de ciblage sont en cours de développement. Sur la base des progrès déjà réalisés, il est prévu que le risque d’événements hors cible puisse être considérablement réduit, voire éliminé, dans un avenir proche pour de nombreuses approches d’édition du génome. Vous trouverez ci-dessous trois approches et les progrès associés.
6.1. Modification de la structure Cas9
Des approches d’ingénierie des protéines peuvent être déployées pour développer de meilleures variantes de Cas9, plus précises et plus efficaces. Sur la base d’études sur la structure CRISPR/Cas9, deux articles récents rapportent des substitutions artificielles telles que la protéine Cas9 résultante présente un taux de non-cible considérablement réduit (Kleinstiver et al., 2016 ; Slaymaker et al., 2016). Les deux études se sont concentrées sur différents domaines de liaison à l’ADN de Cas9, mais ont adopté la stratégie commune consistant à réduire l’affinité relative pour la liaison non spécifique à l’ADN. Cela a remarquablement amélioré la spécificité de la découpe Cas9 sans évidemment nuire à son efficacité globale. Si ces tentatives sont encourageantes, d’autres stratégies seront sans doute à venir et devraient encore améliorer le processus, basées sur la connaissance de la structure CRISPR/Cas9 (Haurwitz et al., 2012 ; Jinek et al., 2014 ; Jore et al., 2011 ; Staals et al., 2013 ; Wiedenheft et al., 2009, 2011).
6.2. Combinaisons techniques de Cas9 avec des sites de clivage modifiés
Cette approche implique l’utilisation de deux coupes d’ADN ciblées, garantissant une meilleure fidélité qu’une seule coupe cible. L’argument de base est que la protéine Cas9 possède deux sites actifs de clivage de l’ADN, impliquant l’acide aspartique, D10, et l’histidine, H840, chacun responsable de couper un seul brin d’ADN, générant ainsi le DSB (Jinek et al., 2014). Sur la base de cette fonctionnalité, il existe deux manières d’inactiver Cas9 : l’inactivation simple et l’inactivation double (Guilinger et al., 2014 ; Ran et al., 2013 ; Tsai et al., 2014). Dans une approche, Cas9 avec une seule inactivation, également appelée Cas9 nickase (Cas9n), dans laquelle un seul des résidus du site actif (D10 ou H840) est remplacé par de l’alanine (A), donne une protéine Cas9 capable de couper un brin. d’ADN double brin. Par conséquent, la fourniture de deux ARN guides qui dirigent la coupe sur des brins opposés à proximité immédiate, médiée par un dimère de coupeurs simples Cas9n, conduit à un DSB efficace et à une stimulation du NHEJ et du HDR pour produire un DSB (voir Figure A-3b). Remarquablement, ces pseudos peuvent être efficaces même à des distances allant jusqu’à ~ 100 pb.
Dans une deuxième approche, les deux sites de clivage de Cas9 sont inactivés, produisant dCas9 déficient en nucléase, qui est ensuite fusionné au domaine de clivage FokI. Comme pour les ZFN et les TALEN, deux monomères Cas9 fournissent une spécificité de reconnaissance pour la dimérisation de FokI et le clivage de l’ADN (voir Figure A-3c). De plus, ces deux stratégies nécessitent des espaceurs de longueur appropriée entre les cibles : si les espaceurs sont trop courts, il y aura deux protéines Cas9 concurrentes, et si les espaceurs sont trop longs, il est plus difficile d’effectuer une découpe efficace. En raison de ces exigences strictes, le taux de non-cible est considérablement réduit, mais avec une difficulté accrue dans la sélection des cibles. Il a été démontré que ces stratégies réduisent considérablement les taux de non-objectifs (Guilinger et al., 2014 ; Ran et al., 2013 ; Tsai et al., 2014).
6.3. Cas9 Base-Editor : édition du génome sans cassures d’ADN double brin
Dans le but d’augmenter l’efficacité et la précision de la réalisation de mutations ponctuelles dans l’ADN génomique, le groupe Liu a récemment développé la variante dite «éditeur de base» de Cas9 (figure 5), qui consiste en une protéine de fusion hautement sophistiquée qui recrute un domaine cytosine désaminase, qui convertit la cytosine en uracile, effectuant ainsi une substitution irréversible de CàT (ou GàA en ciblant le brin complémentaire) sans clivage double brin du squelette de l’ADN (Komor et al., 2016).
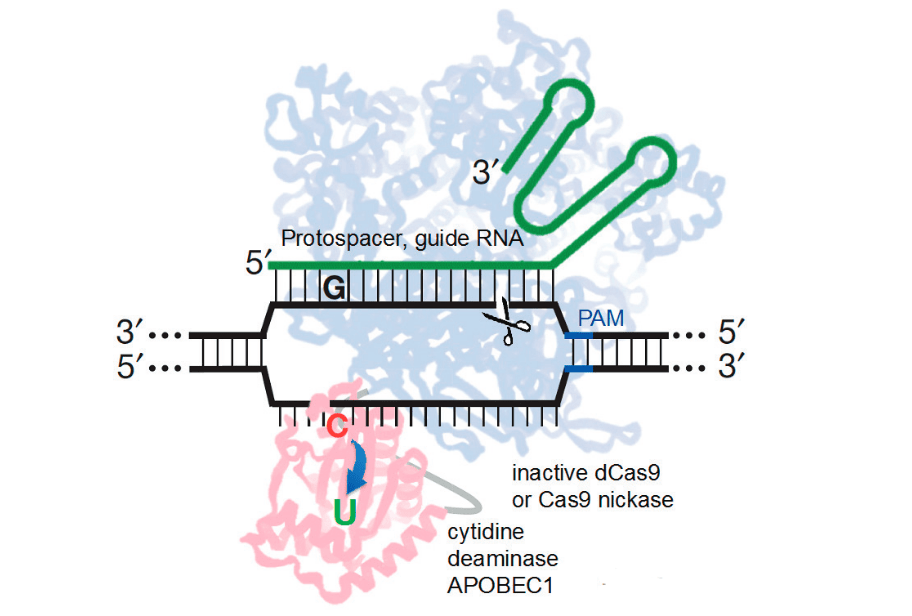
Étant donné que la cytosine désaminase n’agit que sur l’ADN simple brin, l’activité de conversion C en U est ciblée sur une petite fenêtre d’environ 5 nucléotides près de l’extrémité 5 ́ de la séquence protospacer spécifiée par l’ARN guide sur le brin d’ADN déplacé. En évitant les DSB, les modèles d’ADN exogènes et les processus stochastiques de réparation de l’ADN, l’édition de bases introduit des mutations ponctuelles dans les cellules de mammifères non modifiées avec une efficacité pouvant atteindre 75 % et avec un rapport de correction de mutation ponctuelle: indels dépassant 20:1. À l’avenir, il sera important de développer des éditeurs de bases alternatifs capables de gérer un plus large éventail de changements génétiques, permettant idéalement la conversion de n’importe quelle base en n’importe quelle autre base dans une fenêtre définie par l’utilisateur. De plus, il sera important d’évaluer de manière robuste la propension du domaine de la cytosine désaminase à introduire des mutations en dehors de la région d’ADN ciblée par l’ARN guide Cas9.
Une stratégie alternative pour améliorer le HDR, appelée «CORRECT» (étapes consécutives de re-guide ou de re-Cas pour effacer les cibles bloquées par CRISPR/Cas), exploite l’observation selon laquelle la précision du HDR est considérablement augmentée en incorporant des mutations silencieuses bloquant CRISPR/Cas. ainsi que les mutations fonctionnelles souhaitées (Paquet et al., 2016). Cela empêche le clivage et la réparation potentielle du NHEJ qui pourraient perturber le succès des produits HDR. Les auteurs montrent qu’en contrôlant l’emplacement de la mutation ponctuelle introduite par rapport au DSB médié par Cas9, ils peuvent modifier l’efficacité de la mutagenèse, générant des altérations hétérozygotes ou homozygotes dans les cellules souches pluripotentes induites par l’homme (hiPSC).
7. Contrôle de la qualité et assurance qualité pour l’édition génique
La spécificité de l’édition du génome est encore plus importante dans les applications cliniques que dans la recherche en laboratoire. En tant que produit médical ou pratique médicale, l’édition du génome doit être sûre, efficace et rentable. L’élaboration d’un cadre réglementaire pour l’édition du génome devra répondre à diverses questions associées à ces exigences. Comme indiqué ci-dessus, même si les progrès techniques feront probablement des événements hors cible un problème gérable, cette considération restera une préoccupation qui doit être prise en compte par les procédures de contrôle qualité (CQ) et d’assurance qualité (AQ). Dans l’édition du génome somatique, il peut être relativement facile de mettre en place des tests ou des procédures pour traiter les changements hors cible, mais il est probable que ces taux varient selon les types de cellules, ce qui nécessitera la mesure des événements hors cible dans chaque type de cellule ciblé. Bien que les détails puissent varier d’un cas à l’autre, le principe général de surveillance visant à garantir la sécurité et l’efficacité doit être mis en œuvre. En revanche, il serait assez difficile de surveiller les embryons s’ils devaient subir une modification. Des tests d’équivalence fonctionnelle doivent être développés et convenus pour servir de mesures de contrôle de la qualité. Des alternatives peuvent être envisagées, telles que l’édition effectuée sur les cellules progénitrices des spermatozoïdes.
8. Utilisation de dCas9 pour réguler la transcription ou pour faire des modifications épigénétiques
Une stratégie alternative pour l’édition du génome implique l’utilisation de variantes catalytiquement mortes de Cas9. Cela donne une protéine programmable de liaison à l’ADN qui est incapable de générer des cassures simple ou double brin et n’entraîne donc généralement aucune modification dans la séquence d’ADN du génome. Cependant, en fusionnant différents domaines effecteurs avec dCas9, il peut être utilisé soit pour activer (CRISPRa) (Gilbert et al., 2014 ; Konermann et al., 2015 ; Perez-Pinera et al., 2013), soit pour désactiver (CRISPRi ) la transcription (Gilbert et al., 2013 ; Qi et al., 2013) ou pour apporter des modifications spécifiques au locus des marques épigénétiques (modifications de la chromatine qui régulent l’expression des gènes). Il est probable que la plupart, sinon la totalité, de ces changements épigénétiques ne pourraient pas être transmis aux générations suivantes, atténuant ainsi certaines des inquiétudes entourant les approches d’édition de la lignée germinale.
De la même manière, la nature transitoire de ces changements limite leur utilité pour corriger les maladies provoquées par des mutations génétiques. Les utilisations possibles d’une telle ingénierie germinale transitoire incluent cependant la capacité à développer des cellules germinales ou la génération in vitro de cellules souches souhaitées ou de cellules différenciées en phase terminale. De plus, la nature transitoire des changements pourrait augmenter le nombre de gènes pouvant être ciblés en toute sécurité. Par exemple, une régulation négative transitoire du corécepteur CCR5 du VIH pourrait protéger contre le passage vertical du VIH, et cette stratégie pourrait être étendue à d’autres récepteurs viraux. De plus, il est possible d’imaginer que des altérations transitoires de l’expression des gènes pourraient conduire à des changements permanents dans le développement d’un embryon, ce qui pourrait améliorer les effets des mutations héréditaires pathogènes.
Étant donné qu’aucun changement permanent et héréditaire n’est apporté à l’individu, l’utilisation de dCas9 pour modifier l’expression des gènes atténue certaines préoccupations éthiques. Néanmoins, à l’heure actuelle, les utilisations potentielles de dCas9 sur les embryons semblent plutôt limitées par rapport aux approches impliquant l’édition de la lignée germinale, et les applications thérapeutiques plus immédiates de dCas9 impliquent probablement une altération somatique de l’expression des gènes.
9. Ciblage du génome chez les animaux transgéniques
Les mutations génétiques peuvent conduire à un développement anormal et à des maladies. Au cours des dernières décennies, une avancée majeure dans l’étude des conséquences des mutations a été le développement de la capacité d’introduire expérimentalement des mutations conçues et ciblées dans les gènes d’organismes tels que les souris, les mouches des fruits et le poisson zèbre, fournissant ainsi un outil important. comprendre les bases moléculaires et génétiques du développement embryonnaire et des maladies. Ces méthodes peuvent également être utilisées pour corriger des gènes défectueux. Avant de décrire l’application des méthodes précises actuelles d’édition du génome, nous résumerons les principales étapes initialement développées pour modifier génétiquement les animaux.
9.1. Insertion aléatoire d’ADN étranger
La manipulation génétique des animaux est à la base d’une grande partie des recherches visant à comprendre le développement embryonnaire et les maladies humaines. Une technologie puissante repose sur la manipulation d’embryons de souris in vitro et leur transfert à une mère adoptive afin de produire des animaux génétiquement modifiés (figure 6).
Grâce à ces techniques, d’abord l’ADN SV40 (Jaenisch et Mintz, 1974) et plus tard des rétrovirus (Jaenisch, 1976) ont été introduits dans les premiers embryons de souris, conduisant à la génération des premières souris transgéniques qui ont transmis l’ADN étranger à la génération suivante selon Attentes mendéliennes. La méthode la plus largement utilisée pour générer des animaux génétiquement modifiés était la microinjection d’ADN dans des œufs fécondés de souris ou de drosophile, conduisant à la production d’un grand nombre de souris ou de mouches transgéniques portant de l’ADN étranger dans leur lignée germinale (Brinster et al., 1981 ; Costantini et Lacy, 1981 ; Gordon et Ruddle, 1981 ; Rubin et Spradling, 1982).
L’intégration aléatoire d’ADN étranger dans le génome d’un animal peut provoquer la perturbation d’un gène endogène conduisant à son inactivation. Dans cette approche de «mutagenèse insertionnelle», l’ADN intégré est utilisé comme marqueur moléculaire pour l’isolement et l’identification du gène muté. Le gène du collagène I a été le premier gène endogène inactivé par mutagenèse insertionnelle rétrovirale, donnant naissance à des souris mutantes dont le phénotype ressemblait à la maladie des os de verre (Schnieke et al., 1983), une maladie majeure du système squelettique provoquée par des mutations dans un gène du collagène. De même, l’injection d’ADN dans le pronoyau du zygote a produit des souris mutantes par mutagenèse insertionnelle (Mahon et al., 1988). Outre la mutagenèse insertionnelle, l’intégration d’un gène dans le génome peut entraîner une activation transcriptionnelle de gènes voisins, ce qui a été largement utilisé pour étudier les conséquences de l’expression transgénique ectopique (Hammer et al., 1984).
Bien que l’intégration dans le génome conduisant à une mutagenèse insertionnelle ou à l’expression ectopique de transgènes soit efficace pour générer des animaux transgéniques, l’approche souffre d’imprévisibilité car l’insertion d’ADN dans le génome est aléatoire et ne permet pas le ciblage de gènes prédéterminés ou une expression transgénique prévisible. .
9.2. Ciblage des gènes dans les cellules souches embryonnaires
Les cellules souches embryonnaires (ES), initialement isolées des blastocystes de souris, sont capables de se différencier en tous les types de cellules du corps (Evans et Kaufman, 1981 ; Martin, 1981). Il était très intéressant que les cellules ES, lorsqu’elles étaient injectées dans un blastocyste porteur de souris, puissent s’intégrer dans l’embryon en développement et contribuer à tous les tissus somatiques et générer des «souris chimériques». Le fait que les cellules étaient capables de contribuer à la lignée germinale, permettant ainsi la dérivation d’animaux à partir des cellules cultivées, était particulièrement important. Ainsi, l’approche a permis la génération de souris porteuses de l’altération d’un gène endogène suite à une manipulation in vitro des cellules ES.
La première souche de souris dérivée d’une cellule ES portant une mutation dans un gène prédéterminé était une souche avec inactivation du gène HPRT, qui est muté chez les patients humains atteints du syndrome de Lesch-Nyhan, un trouble mental grave. L’isolement des cellules ES mutantes HPRT était simple à l’aide d’un milieu de culture (HAT) qui tue les cellules normales et sélectionne les cellules portant un gène HPRT inactivé (Kuehn et al., 1987). Cette approche sélective, bien que réussie pour HPRT, ne peut pas être utilisée pour modifier d’autres gènes.
9.3. Recombinaison homologue
La découverte de la recombinaison homologue a représenté une avancée majeure car elle a permis l’édition de n’importe quel gène (Doetschman et al., 1987 ; Thomas et Capecchi, 1987). Le ciblage des gènes nécessite la génération d’un vecteur de ciblage contenant des segments d’ADN homologues aux séquences du gène endogène flanquant la modification souhaitée (figure 1). Le vecteur est transfecté dans des cellules ES et les clones correctement ciblés sont sélectionnés (figure 7a). Les cellules portant la modification souhaitée sont injectées dans des blastocystes de souris pour générer des souris chimériques (figure 7b), qui sont croisées avec des souris normales pour obtenir une progéniture portant l’allèle mutant (figure 7c). La recombinaison homologue en combinaison avec les cellules ES a permis aux scientifiques de créer efficacement des souris transmettant des mutations génétiques spécifiques à la génération suivante.
Suite à la génération initiale de souris portant des mutations ciblées de la β2-microglubuline et du gène c-Abl (Schwartzberg et al., 1989 ; Zijlstra et al., 1989), la recombinaison homologue dans les cellules ES est devenue un outil largement utilisé pour l’étude du développement des mammifères et la génération de modèles animaux de maladies génétiques humaines (Solter, 2006). Étant donné que les cellules ES compétentes pour les chimères n’étaient disponibles que dans le système murin, l’édition de gènes par recombinaison homologue était limitée aux souris et ne pouvait pas être facilement utilisée chez d’autres espèces.
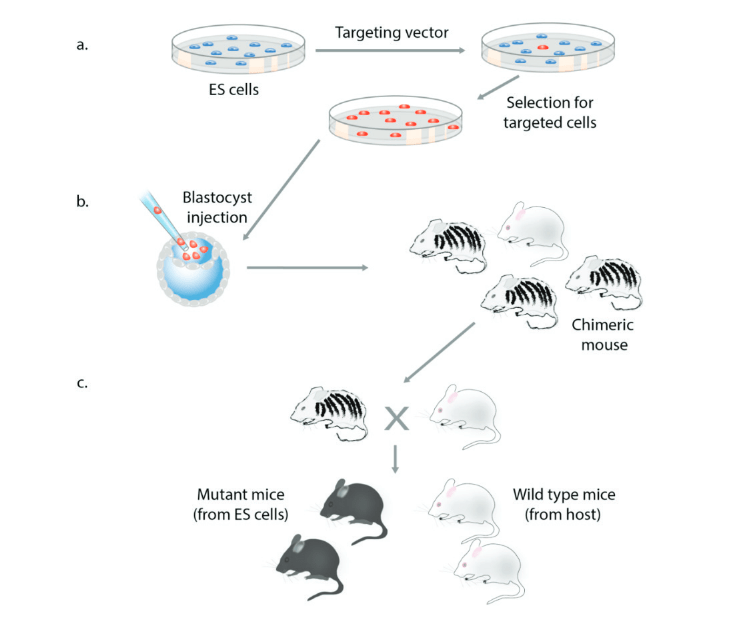
9.4. Clonage nucléaire et génération d’animaux mutants
Le transfert d’un noyau somatique dans un œuf énucléé réinitialise l’état épigénétique du noyau à un état embryonnaire et permet la génération d’animaux tels que Dolly, le premier mammifère cloné (Wakayama et al., 1998 ; Wilmut et al., 1997). La production d’animaux à partir de cellules somatiques par clonage nucléaire a permis la génération d’animaux mutants dans des espèces pour lesquelles aucune cellule ES n’était disponible. La première application réussie du clonage nucléaire en combinaison avec la recombinaison homologue pour produire des animaux de ferme génétiquement modifiés a utilisé des fibroblastes de mouton. Le gène de la 1-antitrypsine humaine a été inséré dans la région 3′ UTR du gène COL1A1, un locus «refuge» pratique donnant une expression prévisible du transgène. Des moutons transgéniques ont été dérivés des fibroblastes ciblés par clonage nucléaire, générant des animaux exprimant la protéine humaine thérapeutiquement importante ; 1-antitrypsine (McCreath et al., 2000).
Les stratégies visant à produire des animaux porteurs d’altérations génétiques artificielles, telles que résumées ci-dessus, reposaient sur la manipulation de cellules ES ou sur le clonage nucléaire, qui demandent tous deux beaucoup de main-d’œuvre et nécessitent des compétences particulières. Cela a radicalement changé lorsque les nouvelles méthodes d’édition du génome basées sur les ZFN, les TALEN et CRISPR/Cas9 sont devenues disponibles (Doudna et Charpentier, 2014). Ces approches, décrites ci-dessus, ont révolutionné la capacité des chercheurs à modifier les gènes de n’importe quelle espèce et à produire des animaux génétiquement modifiés avec une fraction de l’effort et du temps nécessaires et avec beaucoup moins de sophistication et de compétences expérimentales nécessaires que celles requises pour la génération de gènes. animaux édités par des stratégies basées sur les cellules ES ou le transfert nucléaire.
10. Édition du génome chez les embryons
La recombinaison homologue dans le ciblage génétique conventionnel est un processus inefficace et nécessite la sélection de clones cellulaires correctement ciblés dans une culture cellulaire. Dans une deuxième étape, le clone de cellule ES ciblé est injecté dans un blastocyste hôte pour créer un animal chimérique qui, dans une troisième étape, est accouplé pour produire l’animal mutant souhaité, un processus qui peut prendre jusqu’à 1 ou 2 ans (comparer à la figure 7). En revanche, le ciblage des gènes par TALEN ou par CRISPR/Cas9 est si efficace qu’aucune sélection pour un ciblage correct n’est nécessaire (Sakuma et Woltjen, 2014), ce qui permet d’obtenir des animaux génétiquement modifiés en une seule étape par manipulation génétique directe des gènes. œuf fécondé (voir la figure A-8).
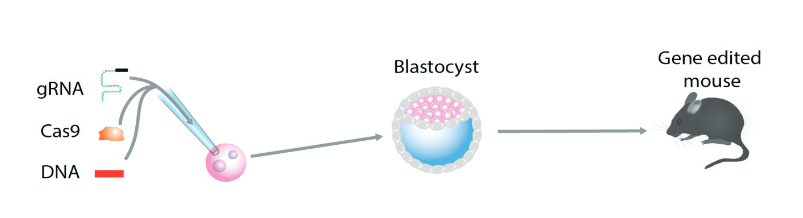
10.1. Édition génétique médiée par CRISPR/Cas9 dans le zygote
L’injection d’ARN guides ainsi que d’ARN Cas9 dans l’œuf fécondé (zygote) a été utilisée pour générer des souris porteuses de mutations dans plusieurs gènes. L’efficacité des DSB d’ADN médiés par Cas9 était élevée et aboutissait à ce que 80 % des ratons portaient des mutations dans les deux allèles de deux gènes différents (Wang et al., 2013). Lorsque les ARN guide et Cas9 ont été co-injectés avec un oligonucléotide porteur d’une mutation ponctuelle, la mutation a été introduite dans deux gènes cibles chez 60 pour cent des ratons. De plus, la génération de mutants conditionnels nécessitant l’insertion de deux sites LoxP dans le même allèle s’est avérée efficace chez le zygote. Ainsi, la mutation médiée par NHEJ ainsi que l’insertion d’ADN au site DSB sont extrêmement efficaces, permettant la génération de souris porteuses de mutations complexes en 3 semaines (le temps de gestation de la souris) au lieu de 1 à 2 ans lors de l’utilisation de cellules ES. ciblage génique médié.
Il a été démontré que la méthode d’édition de gènes CRISPR/Cas9 fonctionne efficacement non seulement chez la souris, mais également chez d’autres espèces, notamment le rat (Li et al., 2013), le poisson zèbre (Hwang et al., 2013), C. elegans (Friedland et al. ., 2013) et la drosophile (Zeng et al., 2015). Il est important de noter que cette approche a permis de générer des primates porteurs de mutations dans des gènes spécifiques (Niu et al., 2014). Plus récemment, deux rapports ont été publiés utilisant l’édition du génome dans des embryons humains préimplantatoires défectueux et ne pouvant donc pas être utilisés pour générer une grossesse (Kang et al., 2016 ; Liang et al., 2015).
Les preuves résumées ci-dessus indiquent que des animaux porteurs de mutations définies dans plusieurs gènes peuvent être générés en une seule étape par manipulation de l’œuf fécondé. Cependant, s’il est destiné à une thérapie génique (correction de gènes mutants), plusieurs complications importantes doivent être prises en compte. Ceux-ci incluent le mosaïcisme fréquent des embryons manipulés, la mutation des deux allèles lorsque l’objectif est de corriger un allèle mutant et l’impossibilité de génotyper l’embryon unicellulaire.
10.2. Mosaïcisme
Le clivage du gène cible et l’insertion de l’ADN au point de cassure double brin peuvent se produire à un stade ultérieur à celui du zygote, par exemple au stade à deux cellules. La conséquence d’une intégration à un stade ultérieur au stade zygote unicellulaire est que la moitié (ou moins, selon le moment de l’insertion de l’ADN) des cellules de l’embryon porteront le gène modifié alors que les autres ne le porteront pas. Les animaux présentant des altérations génétiques dans seulement un sous-ensemble de cellules sont désignés sous le nom de «mosaïques». Les preuves disponibles indiquent que l’incidence du mosaïcisme peut atteindre 50 pour cent ou plus (Wang et al., 2013). L’incidence élevée du mosaïcisme a une conséquence pratique importante : le diagnostic génétique préimplantatoire (DPI) – la biopsie d’une ou de quelques cellules de l’embryon manipulé – ne peut pas être utilisé pour déterminer si le ciblage génétique a abouti à la mutation souhaitée, car les cellules biopsiées peut ne pas refléter le génotype des autres cellules de l’embryon.
10.3. Mutation de l’allèle de type sauvage par clivage médié par Cas9
Le clivage par Cas9 est significativement plus efficace que l’insertion d’un ADN donneur au site de clivage par recombinaison homologue. Cela pose une complication si l’objectif de l’édition génétique chez les embryons est la correction d’un allèle mutant. Pour corriger une mutation donnée, un ARN guide et une construction cible d’ADN sont injectés dans l’embryon. Alors que l’ADN s’intégrera dans l’allèle mutant au niveau du DSB et corrigera la mutation, l’autre allèle sera souvent clivé, créant une nouvelle mutation par NHEJ. Compte tenu de la technologie actuelle, cela pose un problème potentiellement sérieux pour la thérapie génique dans la mesure où l’allèle mutant est corrigé, mais un nouvel allèle mutant est créé. L’inhibition de la réparation par jonction des extrémités à l’aide d’une petite molécule peut aider à atténuer ce problème, car il a été démontré que cela favorise l’insertion de l’ADN par HDR par rapport à celle par NHEJ (Maruyama et al., 2015). Cependant, la mutation indésirable de l’allèle normal reste actuellement une complication de la correction génique médiée par CRISPR/Cas9.
10.4. Génotypage de l’embryon unicellulaire
L’édition du génome des embryons dans le but de corriger un allèle mutant se heurte à un autre problème: comment distinguer un embryon sauvage d’un embryon mutant. Si un parent est porteur d’un gène mutant dominant, 50 pour cent des embryons seront affectés et 50 pour cent seront de type sauvage, et si les deux parents sont porteurs d’une mutation récessive, 75 pour cent des embryons seront normaux et 25 pour cent seront affectés. Puisqu’il n’est pas possible d’utiliser les tests moléculaires actuels pour distinguer les embryons mutants des embryons normaux au stade zygote, toute tentative d’édition de gènes ciblera (et modifiera) une grande fraction des embryons normaux. Il est peu probable qu’un progrès technologique puisse résoudre ce dilemme dans un avenir proche.
11. Le forçage génétique: un mécanisme permettant de propager les altérations génétiques à travers des populations à reproduction sexuée
Il a été proposé que les «endonucléases de référence» naturelles pourraient provoquer la propagation rapide des allèles mutants à travers des populations se reproduisant sexuellement par un processus appelé « forçage génétique » (Burt, 2003, 2014). La propagation d’un tel allèle mutant à travers une population ne nécessite pas d’avantage sélectif pour le porteur du gène mutant mais se propagerait plutôt comme un «gène égoïste» (Esvelt et al., 2014 ; Oye et al., 2014).
Récemment, un vecteur codant à la fois pour Cas9 et un ARN guide a été introduit dans un locus génomique de la drosophile qui régit la couleur de la cuticule, créant ainsi une mutation knock-out. Le clivage médié par Cas9 au cours du développement de la lignée germinale a stimulé la copie de l’insertion dans le locus de type sauvage, de sorte que tous les gamètes femelles portaient l’insertion (figure 9). Il est important de noter que lorsque ces œufs ont été fécondés, la mutation chez les animaux hétérozygotes résultants a converti l’allèle de type sauvage en un allèle mutant au cours de la méiose par clivage du gène cible médié par Cas9, suivi d’un HDR entraînant une mutation homozygote. Ainsi, le vecteur portant le gène Cas9 et un ARN guide, une fois intégré dans un allèle, a conduit à la conversion de l’autre allèle pendant la méiose avec une efficacité de 98 pour cent, provoquant la propagation rapide de l’allèle mutant à travers la population (Gantz et Bier, 2015). Ce processus autocatalytique a été surnommé «réaction mutagène en chaîne».
Si un vecteur portant des séquences corrigeant une mutation donnée en plus du Cas9 et de l’ARN guide était inséré dans un allèle, ces séquences pourraient servir de matrice pendant la méiose et convertir l’autre allèle par recombinaison homologue aboutissant à deux allèles réparés (figure 9a). De même, si un vecteur portant des séquences codant pour un autre gène en plus du Cas9 et de l’ARN guide était intégré dans un allèle, le clivage de l’allèle de type sauvage médié par Cas9 pendant la méiose transférerait le gène «cargo» exogène dans l’autre allèle. , conduisant à des animaux transgéniques homozygotes (Esvelt et al., 2014). Ainsi, les mécanismes de forçage génétique peuvent propager et diffuser efficacement des allèles mutants ou des gènes nouvellement insérés dans les populations animales (figure 9b).
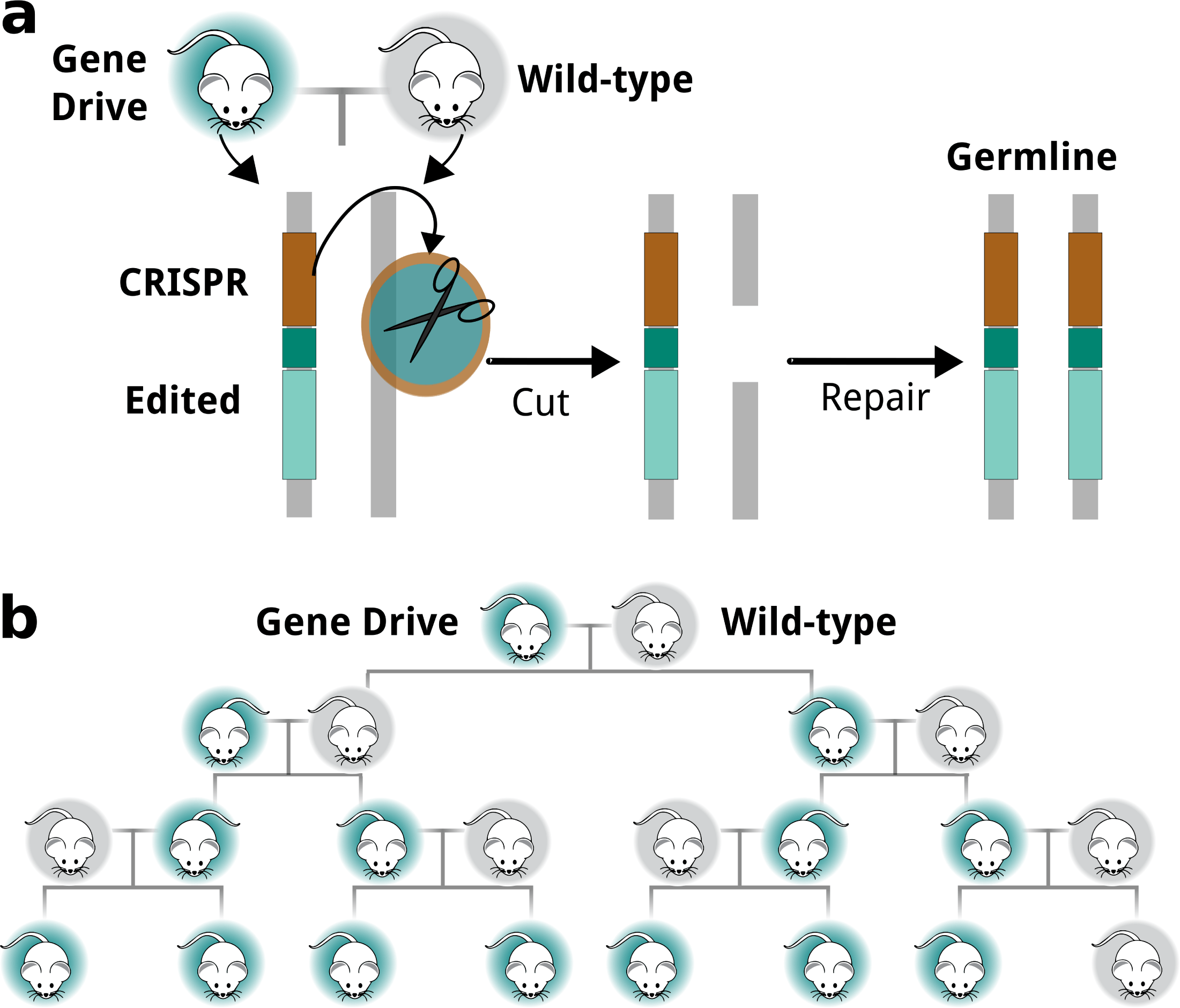
Jusqu’à présent, il a été démontré que les constructions génétiques se propagent à travers les populations d’insectes et ont été proposées pour être utilisées dans le contrôle des populations de moustiques. Il n’a pas encore été démontré que le forçage génétique se comporte de la même manière chez les espèces de mammifères. Cependant, étant donné l’efficacité de l’édition du génome chez les embryons de mammifères, il est possible, voire probable, que les mécanismes de forçage génétique sous-jacents soient également efficaces chez les mammifères et puissent en principe créer des modifications génétiques qui pourraient se propager dans la population. Cependant, étant donné la durée des générations et les modes de reproduction des humains, une telle application du forçage génétique dans l’espèce humaine nécessiterait un nombre d’années démesuré et semble inconcevable.
12. Voies alternatives à l’édition de la lignée germinale héréditaire
L’efficacité et la précision des approches d’édition de gènes CRISPR/Cas9 ont soulevé la possibilité qu’une édition précise du génome soit possible dans des cellules susceptibles de contribuer à la lignée germinale de l’espèce humaine. Pour qu’une altération génétique soit transmise à la génération suivante, elle doit être produite dans (1) les cellules progénitrices pouvant donner naissance aux gamètes (ovules et spermatozoïdes), (2) les ovules et les spermatozoïdes eux-mêmes, ou (3) dans le zygote fécondé ou l’embryon précoce, lorsque toutes les cellules peuvent encore contribuer à la future lignée germinale.
Comme indiqué dans la section précédente, les méthodes d’édition de la lignée germinale ont été plus développées chez la souris et appliquées à un certain nombre d’autres espèces de mammifères, notamment en relation avec les besoins agricoles ou la génération de modèles précliniques de maladies génétiques humaines. Jusqu’à l’avènement récent d’outils avancés d’édition du génome, les altérations des gènes germinaux chez la souris étaient principalement obtenues via l’introduction non ciblée d’ADN transgénique dans le génome du zygote ou par mutagenèse ciblée dans les cellules ES. Cette dernière approche impliquait la recombinaison homologue de vecteurs de ciblage dans le génome de l’hôte, la sélection des clones correctement ciblés et la génération d’animaux chimériques transmettant la lignée germinale (voir les détails dans la section ci-dessus sur l’altération génétique de la lignée germinale). Bien que cette approche se soit révélée extraordinairement puissante pour produire des souris knock-out, des mutations conditionnelles, des lignées rapporteuses et une variété de modèles de maladies humaines, elle reste relativement inefficace et n’a pas été facilement applicable aux altérations génomiques ciblées directes chez les zygotes. Ainsi, l’idée d’éventuelles altérations ou corrections génétiques ciblées sur les embryons humains n’était pas à l’étude.
Cependant, plus récemment, l’efficacité de CRISPR/Cas dans le ciblage de l’édition améliorée par les nucléases sur des sites spécifiques du génome a ouvert de nouvelles perspectives, notamment l’édition possible de la lignée germinale humaine. Peu de temps après la démonstration que CRISPR/Cas pouvait cibler très efficacement des sites spécifiques du génome des cellules de mammifères, il a été démontré que cette approche pouvait être appliquée directement au zygote de souris sans avoir besoin de l’étape intermédiaire des cellules ES. Ainsi, il est devenu possible d’envisager l’édition du génome directement dans les embryons humains. Les seules publications à ce jour sur les tests de CRISPR sur des zygotes humains ont démontré que des mutations ciblées pouvaient être générées, mais ont également démontré que les mutations résultantes étaient souvent complexes et que seuls certains embryons ou cellules embryonnaires étaient porteurs de l’événement ciblé (Kang et al., 2016 ; Liang et coll., 2015). Comme expliqué ci-dessus, ces problèmes intrinsèques à l’édition CRISPR rendent très difficile le concept d’utilisation de l’édition du génome du zygote pour corriger une maladie génétique humaine.
La modification du génome de l’embryon n’est cependant pas le seul moyen potentiel de modifier le génome de la lignée germinale. Les approches qui modifient directement le génome des gamètes – les ovules et les spermatozoïdes – avant la fécondation surmonteraient les problèmes de mosaïcisme et permettraient potentiellement une présélection du gamète correctement ciblé avant la fécondation in vitro.
13. Édition des gènes des gamètes : état actuel
Il existe un certain nombre de voies potentielles d’édition génétique des gamètes, dont certaines sont déjà utilisées chez la souris et d’autres restent à développer.
13.1. Introduction directe de facteurs d’édition dans les ovocytes
Chez la souris, il a été démontré que la nucléase Cas9 héritée de la mère constitue un moyen très efficace de générer des altérations ciblées dans les zygotes résultants (Sakurai et al., 2016), probablement en raison de la disponibilité immédiate de l’enzyme. Bien qu’une telle approche ne soit bien sûr pas applicable aux humains, elle suggère que le fait de précharger les ovocytes ovulés avec les facteurs d’édition avant la fécondation in vitro pourrait être un moyen d’éviter l’édition en mosaïque et d’améliorer l’efficacité. Reste à savoir si cela pourrait réellement favoriser le ciblage des gènes dans le génome de l’ovocyte plutôt qu’après la fécondation. La présélection d’ovocytes mutés ou corrigés serait encore un défi, mais la réduction du mosaïcisme permettrait d’envisager le DPI pour identifier des embryons correctement ciblés.
13.2. Modification des gènes dans le sperme in vitro
Le transfert de gènes médié par les spermatozoïdes est une voie de transgenèse assez bien établie, bien qu’inefficace, chez un certain nombre d’espèces, du poisson au porc (Lavitrano et al., 2013). Ainsi, il devrait être possible d’introduire les composants des systèmes d’édition du génome dans le sperme et de les transporter dans le zygote pour y favoriser l’édition du génome. Plus intéressante est la possibilité d’une édition directe du génome dans les noyaux des spermatozoïdes. Étant donné que les spermatozoïdes sont des cellules qui ne se divisent pas, seule l’édition génétique médiée par NHEJ serait actuellement possible, bien que le mécanisme de réparation soit probablement différent de celui des cellules somatiques (Ahmed et al., 2010). Jusqu’à présent, la correction ou l’altération des gènes par recombinaison homologue n’était possible que dans les cellules en division. Cependant, certains éléments indiquent que ce blocage peut être surmonté (Orthwein et al., 2015).
Le groupe d’Izpisua Belmonte a récemment développé une méthode d’inactivation de gènes basée sur NHEJ : l’intégration de gènes ciblée indépendante de l’homologie (HITI). HITI permet l’inactivation directe de séquences d’ADN au niveau de loci génomiques spécifiques dans les cellules post-mitotiques, par exemple les neurones (Suzuki et al., 2016b). HITI pourrait ouvrir de nouvelles voies à l’édition génétique des spermatozoïdes (et même des ovocytes). L’insertion d’un rapporteur fluorescent transitoire pour identifier les spermatozoïdes porteurs des facteurs d’édition pourrait aider à enrichir les spermatozoïdes potentiellement modifiés par des gènes. La confirmation finale des embryons correctement édités après fécondation in vitro ou injection intracytoplasmique de spermatozoïdes (ICSI) nécessiterait un DPI.
13.3. Modification des gènes dans les cellules souches germinales
Il existe un intérêt biologique et clinique considérable à générer des gamètes à partir de lignées de cellules souches qui peuvent se propager indéfiniment in vitro. Des cellules souches spermatogoniales (SSC) ont été isolées de testicules de souris et ont la capacité de régénérer les spermatozoïdes compétents en matière de fécondation lorsqu’elles sont retransplantées dans le testicule adulte appauvri en cellules germinales (Kanatsu-Shinohara et Shinohara, 2013). L’édition génétique dans les SSC permettrait la présélection de lignées clonales présentant des mutations ciblées appropriées et la possibilité de présélectionner des effets hors cible ou d’autres altérations génomiques ou épigénomiques indésirables avant de générer des gamètes. La preuve de principe d’une telle approche a été publiée (Wu et al., 2015), dans laquelle les auteurs ont corrigé une mutation génétique provoquant des cataractes chez la souris par édition CRISPR/Cas9 dans les SSC. Les SSC ont été retransférées dans les testicules et les spermatides rondes ont été collectées pour l’ICSI. La progéniture a été correctement éditée avec une efficacité de 100 pour cent.
Traduire ce travail chez l’homme comporte de nombreux défis. Bien que des cellules de type SSC aient été isolées de testicules humains (Wu et al., 2015), des lignées cellulaires stables et auto-renouvelables n’ont pas encore été obtenues. Si ce défi est surmonté, il reste encore le défi de générer des gamètes compétents en ICSI à partir des SSC. Chez la souris, cela est réalisé par transfert dans le testicule appauvri en cellules germinales, ce qui n’est pas une solution facile chez l’homme. D’autres approches incluent la génération d’un «testicule reconstitué» avec des SSC mixtes et des cellules de soutien du testicule et sa transplantation sous la capsule testiculaire. Cette approche serait également éthiquement difficile chez les humains. L’utilisation possible de reconstitutions interspécifiques et de transplantations chez des souris immunodéficientes poserait ses propres défis scientifiques et éthiques. La meilleure solution serait de promouvoir la différenciation des SSC en gamètes haploïdes matures dans un système de culture entièrement défini in vitro, un défi qui n’a encore été atteint dans aucun système.
Bien que la possibilité d’appliquer des approches similaires à la lignée germinale femelle soit séduisante, les preuves de l’existence de cellules souches oogoniales sont controversées (Johnson et al., 2004). La plupart des preuves suggèrent qu’il existe une ressource limitée en ovocytes dans l’ovaire des mammifères adultes (Eggan et al., 2006) et aucune preuve de cellules souches endogènes.
13.4. Édition génétique dans les cellules souches pluripotentes suivie de la différenciation des cellules germinales
Les cellules souches embryonnaires pluripotentes ou les cellules souches pluripotentes induites peuvent être générées à partir d’hommes et de femmes, se prêtent facilement à l’édition CRISPR et peuvent être différenciées tout au long du chemin vers des cellules germinales méiotiquement compétentes. Chez la souris, les rapports les plus fiables sur la génération de cellules germinales à partir de cellules ES proviennent de l’imitation des voies connues qui induisent des cellules germinales primordiales à partir de l’épiblaste pluripotent de l’embryon précoce.
Grâce à cette approche, le laboratoire de Saitou a généré des cellules ressemblant à des cellules germinales primordiales (PGC-LC) à partir de cellules ES mâles et femelles. Lorsque les PGC-LC ont été reconstituées avec des cellules de soutien provenant respectivement du testicule ou de l’ovaire et transplantées dans l’environnement du testicule ou de l’ovaire, les chercheurs ont pu récupérer des spermatides ou des ovocytes qui pourraient être utilisés pour générer une progéniture viable lorsqu’ils étaient combinés avec des ovules et des spermatozoïdes normaux (Hayashi et al., 2011, 2012). Des progrès récents ont encore étendu cette approche, soit par coculture des PGC-LC avec des cellules testiculaires en culture pour générer des cellules ressemblant à des spermatides in vitro (Zhou et al., 2016), soit par dérivation de SSC dans des cocultures de cellules souches embryonnaires pluripotentes. avec les cellules somatiques des testicules et la maturation ultérieure en spermatides dans les testicules adultes (Ishikura et al., 2016). Dans les deux cas, des spermatides ont été dérivées, capables de féconder les ovocytes après ICSI et de générer une progéniture viable.
Certaines inquiétudes subsistent quant à savoir si la reprogrammation épigénétique serait complète dans ce système culturel, mais les résultats globaux sont tout à fait remarquables. Un autre développement intéressant vient des groupes Izpisua Belmonte, Okuda et Matsui, qui ont montré que l’inactivation ou l’inactivation de Max dans les cellules souches embryonnaires de souris (CSE) active fortement l’expression de gènes liés aux cellules germinales et entraîne de profonds changements cytologiques pour ressembler à des cellules. subissant une division méiotique (Maeda et al., 2013; Suzuki et al., 2016a). Reste à savoir si des cellules haploïdes fonctionnelles peuvent être générées à l’aide de cette approche.
Ces résultats dans les systèmes murins suscitent l’espoir que des gamètes haploïdes humains pourraient être générés à partir de cellules pluripotentes humaines, avec des implications pour la compréhension de la gamétogenèse et des causes de l’infertilité et offrant potentiellement de nouvelles voies de reproduction dans les couples infertiles. Cela ouvrirait également la voie à la modification génétique des cellules souches pour réparer les causes génétiques connues de l’infertilité ou pour réparer les mutations génétiques dominantes. Cependant, à ce jour, les gamètes humains n’ont pas été générés avec succès à partir de cellules souches pluripotentes, bien que deux articles récents rapportent la génération de PGC-LC précoces à partir de cellules ES humaines (Irie et al., 2015 ; Sasaki et al., 2015). Ces études ont révélé des similitudes et des différences par rapport à la voie de différenciation des cellules germinales de souris. Cela suggère que davantage de connaissances sur la façon dont les cellules germinales se développent réellement dans l’embryon humain, ou peut-être chez le primate non humain, par rapport à l’embryon de souris sont nécessaires pour faire avancer cette recherche.
13.5. Édition de gènes dans les cellules haploïdes ES
La plupart des animaux sont diploïdes et les cellules haploïdes naturelles se limitent généralement aux cellules germinales matures. Récemment, des cellules ES haploïdes androgénétiques (mâles) et parthénogénétiques (femelles) (CSEha) ont été dérivées chez des souris et des rats (Leeb et Wutz, 2011 ; Li et al., 2012, 2014 ; Yang et al., 2012). Les haESC ne contiennent qu’une seule copie des gènes alléliques des cellules diploïdes et se prêtent à une modification génétique avec des approches traditionnelles de ciblage génétique et avec de nouvelles stratégies d’édition du génome basées sur les nucléases (Li et al., 2012, 2014).
Plus intéressant encore, les haESC androgénétiques, qui contiennent un chromosome Y plutôt qu’un chromosome X, peuvent produire une progéniture viable et fertile après injection intracytoplasmique dans des ovocytes matures (Li et al., 2012, 2014). Il a également été démontré que les CESh parthénogénétiques haploïdes de souris sont capables de produire des souris fertiles lorsqu’elles sont injectées dans des ovocytes à la place du génome maternel (Wan et al., 2013). Les deux stratégies peuvent être utilisées pour introduire des modifications génétiques dans la descendance. Plus récemment, des CSEh humaines parthénogénétiques ont également été générées avec succès (Sagi et al., 2016). Les CSEh androgénétiques humaines n’ont pas encore été signalées.
Il existe plusieurs limites aux haESC. Premièrement, le phénotype haploïde s’est révélé instable en culture. Les CSEh subissent une auto-diploïdisation spontanée et nécessitent plusieurs cycles de purification haploïde par tri cellulaire activé par le flux avant de devenir stables en culture. En outre, il existe un manque de CSEha androgénétiques contenant le chromosome Y (Li et al., 2012). Cela est dû au faible potentiel de développement des embryons androgénétiques dotés de chromosomes YY (Latham et al., 2000 ; Tarkowki, 1977). Par conséquent, seules les femelles peuvent actuellement être créées. Avec une reproduction plus poussée, des mâles peuvent alors être obtenus. Un autre inconvénient majeur est que l’efficacité des CSEh androgénétiques à féconder un œuf est très faible (moins de 5 pour cent chez la souris et moins de 2 pour cent chez le rat).
En résumé, bien que la génération de gamètes humains « artificiels » à partir de lignées de cellules souches ne soit pas réalisable actuellement, les travaux sur la souris suggèrent que cela sera probablement possible. La reconstitution de l’environnement des testicules ou des ovaires in vitro peut être réalisée en dérivant à la fois des cellules germinales et des cellules de soutien, telles que des cellules de Sertoli, et des cellules de la granulosa à partir d’une différenciation in vitro de cellules ES humaines. Une meilleure compréhension des voies de signalisation endogènes qui favorisent le développement des cellules germinales et la maturation méiotique facilitera la future dérivation de gamètes humains in vitro. De telles cellules seront immédiatement utiles pour comprendre la gamétogenèse et disséquer les problèmes de fertilité, mais les problèmes de sécurité devront être surmontés avant de pouvoir être utilisées pour la reproduction humaine, avec ou sans modification du génome. La lignée germinale est généralement considérée comme quelque peu protégée des dommages génétiques, contrairement à celle des cellules somatiques, et subit également un remodelage épigénétique important avant la fin de la gamétogenèse. Les deux aspects devraient être répliqués dans les gamètes artificiels générés in vitro.
14. Édition du génome mitochondrial
Les maladies mitochondriales sont un groupe de maladies causées par un dysfonctionnement des mitochondries dû à des mutations de l’ADN mitochondrial (ADNmt). Les maladies mitochondriales sont associées à la dégénérescence des tissus et des organes qui ont des besoins énergétiques élevés – notamment les muscles, le cœur et le cerveau – qui conduisent, entre autres pathologies, à des myopathies, des cardiomyopathies, des neuropathies, des encéphalopathies, une acidose lactique, un syndrome semblable à un accident vasculaire cérébral. et la cécité (Taylor et Turnbull, 2005). Le pourcentage de molécules d’ADNmt mutées détermine généralement si un patient est symptomatique ou non. Actuellement, il n’existe aucun remède contre les maladies mitochondriales, et pour les patients suffisamment en bonne santé pour avoir des enfants, le conseil génétique et le DPI représentent les meilleures options pour prévenir la transmission des maladies. Cependant, en raison de l’hérédité non mendélienne de l’ADNmt et des niveaux d’hétéroplasmie potentiellement différents entre les différents blastomères, le DPI ne peut que réduire, et non éliminer, le risque de transmission de la maladie.
Les techniques de remplacement mitochondrial récemment développées impliquent une série de manipulations techniques complexes du génome nucléaire entre les ovocytes de la patiente et du donneur qui aboutissent à la génération d’embryons porteurs de matériel génétique de trois origines différentes (Paull et al., 2012 ; Tachibana et al., 2012). Pour ces raisons, les techniques de remplacement mitochondrial ont soulevé des préoccupations biologiques, médicales et éthiques (Hayden, 2013 ; Reinhardt et al., 2013). Les techniques de remplacement mitochondrial ont de faibles taux de réussite, et des études sur des organismes inférieurs ont signalé des problèmes potentiels résultant de l’incompatibilité entre l’ADN nucléaire et l’ADNmt lors du remplacement mitochondrial (Reinhardt et al., 2013).
Une nouvelle approche thérapeutique alternative a été récemment développée pour éliminer l’ADNmt muté dans la lignée germinale. Grâce à une endonucléase ciblée sur les mitochondries, la transmission de l’ADNmt ciblé dans la lignée germinale de souris à la génération suivante a été évitée (Reddy et al., 2015). En raison du nombre limité de mutations de l’ADNmt qui peuvent être ciblées par les endonucléases de restriction, des efforts ont été déployés pour cibler la plupart des mutations de l’ADNmt à l’aide de nucléases effectrices de type activateur de transcription (mito-TALEN) et de ZFN ciblées sur les mitochondries (Bacman et al., 2013 ; Gammage et al., 2014). Les mito-TALEN ont pu éliminer spécifiquement l’ADNmt ciblé dans la lignée germinale de souris (Reddy et al., 2015).
Il est important de noter que la technique d’injection de nucléases (par exemple, les mito-TALEN) dans des ovocytes ou des embryons précoces implique une simple micro-injection d’ARNm qui code pour les nucléases. De plus, l’utilisation du signal de localisation mitochondriale (par exemple Cox8 et ATP5b) limite la translocation aux seules mitochondries. Une mise en garde concernant cette technologie est que l’élimination de niveaux élevés d’ADNmt muté dans les ovocytes conduira à la génération d’embryons avec un faible nombre d’ADNmt normal qui, s’ils ne se répliquent pas après l’implantation, pourraient entraîner une fausse couche. Le DPI pourrait être utilisé pour la sélection et le transfert d’embryons contenant des niveaux plus élevés d’ADNmt normal. Il est important de noter que, contrairement à l’édition nucléaire, l’édition de l’ADNmt ne vise pas à corriger les mutations, mais à éliminer l’ADN muté, ce qui est possible en raison de la présence de plusieurs copies d’ADNmt dans les ovocytes.
De plus, en raison de la très faible activité des mécanismes de réparation dans les mitochondries, la fréquence de religature de l’ADNmt cible et l’introduction de nouvelles mutations seraient très rares. En outre, des outils d’édition mitochondriales similaires pourraient à l’avenir également être utilisés pour éliminer l’ADNmt muté dans les gamètes dérivés de cellules souches. Enfin, une combinaison d’outils d’édition de gènes mitochondriaux avec des techniques de remplacement mitochondrial pourrait représenter une option alternative, à l’avenir, pour empêcher la transmission germinale de mutations dans l’ADNmt responsables non seulement de maladies mitochondriales spécifiques, mais également de situations où des altérations de la fonction mitochondriale contribue à des pathologies telles que le cancer, le diabète et les maladies associées au vieillissement.
Références
- Ahmed, E. A., P. de Boer, M. E. P. Philippens, H. B. Kal, and D. G. de Rooij. 2010. Parp1- XRCC1 and the repair of DNA double strand breaks in mouse round spermatids. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 683(1-2):84-90.
- Auer, T. O., K. Duroure, A. De Cian, J. P. Concordet, and F. Del Bene. 2014. Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Research 24(1):142-153.
- Bacman, S. R., S. L. Williams, M. Pinto, S. Peralta, and C. T. Moraes. 2013.Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nature Medicine 19(9):1111-1113.
- Barrangou, R., C. Fremaux, H. Deveau, M. Richards, P. Boyaval, S. Moineau, D. A. Romero, and P. Horvath. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819):1709-1712.
- Bibikova, M., D. Carroll, D. Segal, J. K. Trautman, J. Smith, Y. G. Kim, and S. Chandrasegaran. 2001. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Molecular and Cellular Biology 21(1):289-297.
- Bibikova, M., M. Golic, M., K. G. Golic, and D. Carroll. 2002. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161(3):1169-1175. Bibikova, M., K. Beumer, J. K. Trautman, and D. Carroll. 2003. Enhancing gene targeting with designed zinc finger nucleases. Science 300(5620):764.
- Brinster, R. L., H. Y. Chen, M. Trumbauer, A. W. Senear, R. Warren, and R. D. Palmiter. 1981. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell 27(1 Pt. 2):223-231.
- Burt, A. 2003. Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proceedings of The Royal Society B: Biological Sciences 270(1518):921-928.
- Burt, A. 2014. Heritable strategies for controlling insect vectors of disease. Philosophical Transactions of the Royal Society B: Biological Sciences 369(1645):20130432.
- Carroll, D. 2014. Genome engineering with targetable nucleases. Annual Review of Biochemistry 83:409-439.
- Chevalier, B. S., T. Kortemme, M. S. Chadsey, D. Baker, R. J. Monnat, and B. L. Stoddard. 2002. Design, activity, and structure of a highly specific artificial endonuclease. Molecular Cell 10(4): 895-905.
- Chiarle, R., Y. Zhang, R. L. Frock, S. M. Lewis, B. Molinie, Y. J. Ho, D. R. Myers, V. W. Choi, M. Compagno, D. J. Malkin, D. Neuberg, S. Monti, C. C. Giallourakis, M. Gostissa, and F. W. Alt. 2011. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147(1):107-119.
- Cho, S. W., S. Kim, J. M. Kim, and J. S. Kim. 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature Biotechnology 31(3):230-232.
- Cong, L., F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini, and F. Zhang. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819-823.
- Costantini, F., and E. Lacy. 1981. Introduction of a rabbit beta-globin gene into the mouse germ line. Nature 294(5836):92-94.
- Cox, D. B. T., R. J. Platt, and F. Zhang. 2015. Therapeutic genome editing: Prospects and challenges. Nature Medicine 21(2):121-131.
- Deltcheva, E., K. Chylinski, C. M. Sharma, K. Gonzales, Y. Chao, Z. A. Pirzada, M. R. Eckert, J. Vogel, and E. Charpentier. 2011. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340):602-607.
- Desjarlais, J. R., and J. M. Berg. 1992. Toward rules relating zinc finger protein sequences and DNA binding site preferences. Proceedings of the National Academy of Sciences of the United States of America 89(16):7345-7349.
- Dickinson, D. J., J. D. Ward, D. J. Reiner, and B. Goldstein. 2013. Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nature Methods 10(10):1028-1034.
- Doetschman, T., R. G. Gregg, N. Maeda, M. L. Hooper, D. W. Melton, S. Thompson, and O. Smithies. 1987. Targeted correction of a mutant HPRT gene in mouse embryonic stem cells. Nature 330:576-578.
- Doudna, J. A., and E. Charpentier. 2014. Genome editing: The new frontier of genome engi- neering with CRISPR-Cas9. Science 346(6213):1258096.
- Eggan, K., S. Jurga, R. Gosden, I. M. Min, and A. J. Wagers. 2006. Ovulated oocytes in adult mice derive from non-circulating germ cells. Nature 441(7097):1109-1114.
- Esvelt, K. M., A. L. Smidler, F. Catteruccia, and G. M. Church. 2014. Concerning RNA-guided gene drives for the alteration of wild populations. eLife e03401.
- Evans, M. J., and M. H. Kaufman. 1981. Establishment in culture of pluripotential cells from mouse embryos. Nature 292(5819):154-156.
- Friedland, A. E., Y. B. Tzur, K. M. Esvelt, M. P. Colaiacovo, G. M. Church, and J. Calarco. 2013. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nature Methods 10(8):741-743.
- Fu, Y., J. A. Foden, C. Khayter, M. L. Maeder, D. Reyon, J. K. Joung, and J. D. Sander. 2013. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology 31(9):822-826.
- Gabriel, R., A. Lombardo, A. Arenas, J. C. Miller, P. Genovese, C. Kaeppel, A. Nowrouzi, C. C. Bartholomae, J. Wang, G. Friedman, M. C. Holmes, P. D. Gregory, H. Glimm, M. Schmidt, L. Naldini, and C. von Kalle. 2011. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nature Biotechnology 29(9):816-823.
- Gammage, P. A., J. Rorbach, A. I. Vincent, E. J. Rebar, and M. Minczuk. 2014. Mitochondri- ally targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Molecular Medicine 6(4):458-466.
- Gantz, V., and E. Bier. 2015. The mutagenic chain reaction: A method for converting heterozygous to homozygous mutations. Science 348(6233):442-444.
- Garneau, J. E., M. E. Dupuis, M. Villion, D. A. Romero, R. Barrangou, P. Boyaval, C. Fremaux, P. Horvath, A. H. Magadan, and S. Moineau. 2010. The CRISPR/Cas bacte- rial immune system cleaves bacteriophage and plasmid DNA. Nature 468(7320):67-71.
- Gasiunas, G., R. Barrangou, P. Horvath, and V. Siksnys. 2012. Cas9-crRNA ribonucleo-protein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America 109(39):E2579-E2586.
- Gilbert, L. A., M. H. Larson, L. Morsut, Z. Liu, G. A Brar, S. E. Torres, N. Stern-Ginossar, O. Brandman, E. H. Whitehead, J. A. Doudna, W. A. Lim, J. S. Weissman, and L. S. Qi. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154(2):442-451.
- Gilbert, L. A., M. A. Horlbeck, B. Adamson, J. E. Villalta, Y. Chen, E. H. Whitehead, C. Guimaraes, B. Panning, H. L. Ploegh, M. C. Bassik, L.S. Qi, M. Kampmann, and J. S. Weissman. 2014. Genome-scale CRISPR-mediated control of gene repression and activa- tion. Cell 159(3):647-661.
- Gordon, J. W., and F. H. Ruddle. 1981. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 214(4526):1244-1246.
- Guilinger, J. P., D. B. Thompson, and D. R. Liu. 2014. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nature Biotechnology 32(6):577-582.
- Hammer, R. E., R. D. Palmiter, and R. L. Brinster. 1984. Partial correction of murine heredi- tary growth disorder by germ-line incorporation of a new gene. Nature 311(5981):65-67.
- Haurwitz, R. E., S. H. Sternberg, and J. A. Doudna. 2012. Csy4 relies on an unusual catalytic dyad to position and cleave CRISPR RNA. The EMBO Journal 31(12):2824-2832.
- Hayashi, K., H. Ohta, K. Kurimoto, S. Aramaki, and M. Saitou. 2011. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146(4):519-532.
- Hayashi, K., S. Ogushi, K. Kurimoto, S. Shimamoto, H. Ohta, and M. Saitou. 2012. Off-spring from oocytes derived from in vitro primordial germ cell-like cells in mice. Science 338(6109):971-975.
- Hayden, E. C. 2013. Regulators weigh benefits of “three-parent” fertilization. Nature 502(7471):284-285.
- Hsu, P. D., D. A. Scott, J. A. Weinstein, F. A. Ran, S. Konermann, V. Agarwala, Y. Li, E. J. Fine, X. Wu, O. Shalem, T. J. Cradick, L. A. Marraffini, G. Bao, and F. Zhang. 2013. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology 31(9):827-832.
- Hwang, W. Y., Y. Fu, D. Reyon, M. L. Maeder, S. Q. Tsai, J. D. Sander, R. T. Peterson, J. R. Yeh, and J. K. Joung. 2013. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology 31(3):227-229.
- Irie, N., L. Weinberger, W. W. Tang, T. Kobayashi, S. Viukov, Y. S. Manor, S. Dietmann, J. H. Hanna, and M. A. Surani. 2015. SOX17 is a critical specifier of human primordial germ cell fate. Cell 160(1-2):253-268.
- Ishikura, Y., Y. Yabuta, H. Ohta, K. Hayashi, T. Nakamura, I. Okamoto, T. Yamamoto, K. Kurimoto, K. Shirane, H. Sasaki, and M. Saitou. 2016. In Vitro Derivation and Propa- gation of Spermatogonial Stem Cell Activity from Mouse Pluripotent Stem Cells. Cell Reports 17(10):2789-2804.
- Jaenisch, R. 1976. Germ line integration and Mendelian transmission of the exogenous Moloney leukemia virus. Proceedings of the National Academy of Sciences of the United States of America 73(4):1260-1264.
- Jaenisch, R., and B. Mintz. 1974. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proceedings of the National Academy of Sciences of the United States of America 71(4):1250-1254.
- Jamieson, A. C., S. H. Kim, and J. A. Wells. 1994. In vitro selection of zinc fingers with altered DNA-binding specificity. Biochemistry 33(19):5689-5695.
- Jiang, W., D. Bikard, D. Cox, F. Zhang, and L. A. Marraffini. 2013. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology 31(3):233-239.
- Jinek, M., K. Chylinski, I. Fonfara, M. Hauer, J.A. Doudna, and E. Charpentier. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816-821.
- Jinek, M., A. East, A. Cheng, S. Lin, E. Ma, and J. Doudna. 2013. RNA-programmed genome editing in human cells. eLife 2:e00471.
- Jinek, M., F. Jiang, D. W. Taylor, S. H. Sternberg, E. Kaya, E. Ma, C. Anders, M. Hauer, K. Zhou, S. Lin, M. Kaplan, A. T. Iavarone, E. Charpentier, E. Nogales, and J. A. Doudna. 2014. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343(6176):1247997.
- Johnson, J., J. Canning, T. Kaneko, J. K. Pru, and J. L. Tilly. 2004. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 428(6979):145-150.
- Jore, M. M., M. Lundgren, E. van Duijn, J. B. Bultema, E. R. Westra, S. P. Waghmare, B. Wiedenheft, Ü. Pul, R. Wurm, R. Wagner, M. R. Beijer, A. Barendregt, K. Zhou, A. P. L. Snijders, M. J. Dickman, J. A. Doudna, E. J. Boekema, A. J. R. Heck, J. van der Oost, and S. J. J. Brouns. 2011. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nature Structural & Molecular Biology 18(5):529-536.
- Joung, J. K., and J. D. Sander. 2013. TALENs: A widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology 14(1):49-55.
- Kanatsu-Shinohara, M., and T. Shinohara. 2013. Spermatogonial stem cell self-renewal and development. Annual Reviews Cell and Developmental Biology 29:163-187.
- Kang, X., W. He, Y. Huang, Q. Yu, Y. Chen, X. Gao, X. Sun, and Y. Fan. 2016. Introducing precise genetic modifications into human 3PN embryos by CRISPR/Cas-mediated genome editing. Journal of Assisted Reproduction and Genetics 33(5):581-588.
- Kim, Y. G., J. Cha, and S. Chandrasegaran. 1996. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences of the United States of America 93(3):1156-1160.
- Kleinstiver, B. P., V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng, and J. K. Joung. 2016. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide offtarget effects. Nature 529(7587):490-495.
- Komor, A. C., Y. B. Kim, M. S. Packer, J. A. Zuris, and D. R. Liu. 2016. Programmable edit- ing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533(7603):420-424.
- Konermann, S., M. D. Brigham, A. E. Trevino, J. Joung, O. O. Abudayyeh, C. Barcena, P. D. Hsu, N. Habib, J. S. Gootenberg, H. Nishimasu, O. Nureki, and F. Zhang. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517(7536):583-588.
- Kuehn, M., A. Bradley, E. Robertson, and M. Evans. 1987. A potential animal model for Lesch-Nyhan syndrome through introduction of HPRT mutations into mice. Nature 326(6110):295-298.
- Latham, K. E., B. Patel, F. D. Bautista, and S. M. Hawes. 2000. Effects of X chromosome number and parental origin on X-linked gene expression in preimplantation mouse embryos. Biology of Reproduction 63(1):64-73.
- Lavitrano, M., R. Giovannoni, and M. G. Cerrito. 2013. Methods for sperm-mediated gene transfer. In Spermatogenesis: Methods and protocols, Vol. 927, edited by D. Carrell and K. I. Aston. New York: Humana Press. Pp. 519-529.
Leeb, M., and A. Wutz. 2011. Derivation of haploid embryonic stem cells from mouse embryos. Nature 479(7371):131-134. - Li, W., L. Shuai, H. Wan, M. Dong, M. Wang, L. Sang, C. Feng, G.Z. Luo, T. Li, X. Li, L. Wang, Q. Y. Zheng, C. Sheng, H. J. Wu, Z. Liu, L. Liu, L. Wang, X. J. Wang, X. Y. Zhao, and Q. Zhou. 2012. Androgenetic haploid embryonic stem cells produce live transgenic mice. Nature 490(7420):407-411.
- Li, W., F. Teng, T. Li, and Q. Zhou. 2013. Simultaneous generation and germline transmis- sion of multiple gene mutations in rat using CRISPR-Cas systems. Nature Biotechnology 31(8):684-686.
- Li, W., X. Li, T. Li, M. G. Jiang, H. Wan, G. Z. Luo, C. Feng, X. Cui, F. Teng, Y. Yuan, Q. Zhou, Q. Gu, L. Shuai, J. Sha, Y. Xiao, L. Wang, Z. Liu, X. J. Wang, X. Y. Zhao, and Q. Zhou. 2014. Genetic modification and screening in rat using haploid embryonic stem cells. Cell Stem Cell 14(3):404-414.
- Liang, P., Y. Xu, X. Zhang, C. Ding, R. Huang, Z. Zhang, J. Lv, X. Xie, Y. Chen, Y. Li, Y. Sun, Y. Bai, Z. Songyang, W. Ma, C. Zhou, and J. Huang. 2015. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein & Cell 6(5):363-372.
- Maeda, I., D. Okamura, Y. Tokitake, M. Ikeda, H. Kawaguchi, N. Mise, K. Abe, T. Noce, A. Okuda, and Y. Matsui. 2013. Max is a repressor of germ cell-related gene expression in mouse embryonic stem cells. Nature Communications 4:1754.
- Mahon, K. A., P. A. Overbeek, and H. Westphal. 1988. Prenatal lethality in a transgenic mouse line is the result of a chromosomal translocation. Proceedings of the National Academy of Sciences of the United States of America 85(4):1165-1168.
- Makarova, K. S., N. V Grishin, S. A Shabalina, Y. I. Wolf, and E. V. Koonin. 2006. A puta- tive RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biology Direct 1(1):7.
- Mali, P., L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville, and G. M. Church. 2013. RNA-guided human genome engineering via Cas9. Science 339(6121): 823-826.
- Martin, G. R. 1981. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Sciences of the United States of America 78(12):7634-7638.
- Maruyama, T., S. K. Dougan, M. C. Truttmann, A. M. Bilate, J. R. Ingram, and H. L. Ploegh. 2015. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nature Biotechnology 33(5):538-542.
- McCreath, K. J., J. Howcroft, K. H. Campbell, A. Colman, A. E. Schnieke, and A. J. Kind. 2000. Production of gene-targeted sheep by nuclear transfer from cultured somatic cells. Nature 405(6790):1066-1069.
- Mojica, F. J. M., C. Diez-Villasenor, J. Garcia-Martinez, and E. Soria. 2005. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. Journal of Molecular Evolution 60(2):174-182.
Naldini, L. 2015. Gene therapy returns to centre stage. Nature 526(7573):351-360. - Niu, Y., B. Shen, Y. Cui, Y. Chen, J. Wang, L. Wang, Y. Kang, X. Zhao, W. Si, W. Li, A. P. Xiang, J. Zhou, X. Guo, Y. Bi, C. Si, B. Hu, G. Dong, H. Wang, Z. Zhou, T. Li, T. Tan, X. Pu, F. Wang, S. Ji, Q. Zhou, X. Huang, W. Ji, and J. Sha. 2014. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156(4):836-843.
- Orthwein, A., S. M. Noordermeer, M. D. Wilson, S. Landry, R. I. Enchev, A. Sherker, M. Munro, J. Pinder, J. Salsman, G. Dellaire, B. Xia, M. Peter, and D. Durocher. 2015. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528(7582):422-426.
- Oye, K. A., K. Esvelt, E. Appleton, F. Catteruccia, G. Church, T. Kuiken, S. B. Lightfoot, J. McNamara, A. Smidler, and J. P. Collins. 2014. Biotechnology: Regulating gene drives. Science 345(6197):626-628.
- Paquet, D., D. Kwart, A. Chen, A. Sproul, S. Jacob, S. Teo, K. M. Olsen, A. Gregg, S. Noggle, and M. Tessier-Lavigne. 2016. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533(7601):125-129.
- Paull, D., V. Emmanuele, K.A. Weiss, N. Treff, L. Stewart, H. Hua, M. Zimmer, D. J. Kahler, R. S. Goland, S. A. Noggle, R. Prosser, M. Hirano, M. V. Sauer, and D. Egli. 2012. Nuclear genome transfer in human oocytes eliminates mitochondrial DNA variants. Nature 493(7434):632-637.
- Perez-Pinera, P., D. D. Kocak, C. M. Vockley, A. F. Adler, A. M. Kabadi, L. R. Polstein, P. T. Thakore, K. A. Glass, D. G. Ousterout, K. W. Leong, F. Guilak, G. E. Crawford, T. E. Reddy, and C. A. Gersbach. 2013. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nature Methods 10(10):973-976.
- Pourcel, C., G. Salvignol, and G. Vergnaud. 2005. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 151(Pt. 3):653-663.
- Qasim, W., H. Zhan, S. Samarasinghe, S. Adams, P. Amrolia, S. Stafford, K. Butler, C. Rivat, G. Wright, and K. Somana. 2017. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science Translational Medicine 9(374):eaaj2013
- Qi, L. S., M. H. Larson, L. A. Gilbert, J. A. Doudna, J. S. Weissman, A. P. Arkin, and W. A. Lim. 2013. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):1173-1183.
- Ran, F. A., P. D. Hsu, C. Y. Lin, J. S. Gootenberg, S. Konermann, A. E. Trevino, D. A. Scott, A. Inoue, S. Matoba, Y. Zhang, and F. Zhang. 2013. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380-1389.
- Rebar, E. J., and C. O. Pabo. 1994. Zinc finger phage: Affinity selection of fingers with new DNA-binding specificities. Science 263(5147):671-673.
- Reddy, P., A. Ocampo, K. Suzuki, J. Luo, S. R. Bacman, S. L. Williams, A. Sugawara, D. Okamura, Y. Tsunekawa, J. Wu, D. Lam, X. Xiong, N. Montserrat, C. R. Esteban, G. H. Liu, I. Sancho-Martinez, D. Manau, S. Civico, F. Cardellach, M. del Mar O’Callaghan, J. Campistol, H. Zhao, J. M. Campistol, C. T. Moraes, and J. C. I. Belmonte. 2015. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 161(3):459-469.
- Reinhardt, K., D. K. Dowling, and E. H. Morrow. 2013. Medicine: Mitochondrial replace- ment, evolution, and the clinic. Science 341(6152):1345-1346.
- Rouet, P., F. Smih, and M. Jasin. 1994. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Molecular and Cellular Biology 14(12):8096-8106.
- Rubin, G. M., and A. C. Spradling. 1982. Genetic transformation of Drosophila with transposable element vectors. Science 218(4570):348-353.
- Sagi, I., G. Chia, T. Golan-Lev, M. Peretz, U. Weissbein, L. Sui, M. V. Sauer, O. Yanuka, D. Egli, and N. Benvenisty. 2016. Derivation and differentiation of haploid human embry- onic stem cells. Nature 532(7597):107-111.
- Sakuma, T., and K. Woltjen. 2014. Nuclease-mediated genome editing: At the front-line of functional genomics technology. Development, Growth & Differentiation 56(1):2-13.
- Sakurai, T., A. Kamiyoshi, H. Kawate, C. Mori, S. Watanabe, M. Tanaka, R. Uetake, M. Sato, and T. Shindo. 2016. A non-inheritable maternal Cas9-based multiple-gene editing system in mice. Scientific Reports 6:20011.
- Sapranauskas, R., G. Gasiunas, C. Fremaux, R. Barrangou, P. Horvath, and V. Siksnys. 2011. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Research 39(21):9275-9282.
- Sander, J. D., and J. K. Joung. 2014. CRISPR-Cas systems for editing, regulating and targeting genomes. Nature Biotechnology 32(4):347-350.
- Sasaki, K., S. Yokobayashi, T. Nakamura, I. Okamoto, Y. Yabuta, K. Kurimoto, H. Ohta, Y. Moritoki, C. Iwatani, H. Tsuchiya, S. Nakamura, K. Sekiguchi, T. Sakuma, T. Yamamoto, T. Mori, K. Woltjen, M. Nakagawa, T. Yamamoto, K. Takahashi, S. Yamanaka, and M. Saitou. 2015. Robust in vitro induction of human germ cell fate from pluripotent stem cells. Cell Stem Cell 17(2):178-194.
- Schnieke, A., K. Harbers, and R. Jaenisch. 1983. Embryonic lethal mutation in mice induced by retrovirus insertion into the a1(I) collagen gene. Nature 304(5924):315-320.
- Schwartzberg, P., S. Goff, and E. Robertson. 1989. Germ-line transmission of a c-Abl mutation produced by targeted gene disruption in ES cells. Science 246(4931):799-803.
- Silva, G., L. Poirot, R. Galetto, J. Smith, G. Montoya, P. Duchateau, and F. Paques. 2011. Meganucleases and other tools for targeted genome engineering: Perspectives and chal- lenges for gene therapy. Current Gene Therapy 11(1):11-27.
- Slaymaker, I. M., L. Gao, B. Zetsche, D. A. Scott, W. X. Yan, and F. Zhang. 2016. Rationally engineered Cas9 nucleases with improved specificity. Science 351(6268):84-88.
- Solter, D. 2006. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell research. Nature Reviews Genetics 7:319-327.
- Staals, R. H., J. Y. Agari, S. Maki-Yonekura, Y. Zhu, D. W. Taylor, E. van Duijn, A. Barendregt, M. Vlot, J. J. Koehorst, K. Sakamoto, A. Masuda, N. Dohmae, P. J. Schaap, J. A. Doudna, A. J. R. Heck, K. Yonekura, J. van der Oost, and A. Shinkai. 2013. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of Thermus ther- mophilus. Molecular Cell 52(1):135-145.
- Suzuki, A., M. Hirasaki, T. Hishida, J. Wu, D. Okamura, A. Ueda, M. Nishimoto, Y. Nakachi, Y. Mizuno, Y. Okazaki, Y. Matsui, J. C. I. Belmonte, and A. Okuda. 2016a. Loss of MAX results in meiotic entry in mouse embryonic and germline stem cells. Nature Communications 7:11056.
- Suzuki, K., Y. Tsunekawa, R. Hernandez-Benitez, J. Wu, J. Zhu, E. J. Kim, F. Hatanaka, M. Yamamoto, T. Araoka, Z. Li, M. Kurita, T Hishida, M. Li, E. Aizawa, S. Guo, S. Chen, A. Goebl, R. D. Soligalla, J. Qu, T. Jiang, X. Fu, M. Jafari, C. R. Esteban, W. T. Berggren, J. Lajara, E. Nuñez-Delicado, P. Guillen, J. M. Campistol, F. Matsuzaki, G. H. Liu, P. Magistretti, K. Zhang, E. M. Callaway, K. Zhang, and J. C. Belmonte. 2016b. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540:144-149.
- Tachibana, M., P. Amato, M. Sparman, J. Woodward, D. M. Sanchis, H. Ma, N. M. Gutierrez, R. Tippner-Hedges, E. Kang, H. S. Lee, C. Ramsey, K. Masterson, D. Battaglia, D. Lee, D. Wu, J. Jensen, P. Patton, S. Gokhale, R. Stouffer, and S. Mitalipov. 2012. Towards germline gene therapy of inherited mitochondrial diseases. Nature 493(7434):627-631.
- Tarkowki, A. K. 1977. In vitro development of haploid mouse embryos produced by bisection of one-cell fertilized eggs. Journal of Embryology and Experimental Morphology 38:187-202.
- Taylor, R. W., and D. M. Turnbull. 2005. Mitochondrial DNA mutations in human disease. Nature Reviews Genetics 6(5):389-402.
- Thomas, K., and M. Capecchi. 1987. Site directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51(3):503-512.
- Tsai, S. Q., and J. K. Joung. 2016. Defining and improving the genome-wide specificities of CRISPR-Cas9 nuclease. Nature Reviews Genetics 17(5):300-312.
- Tsai, S. Q., N. Wyvekens, C. Khayter, J.A. Foden, V. Thapar, D. Reyon, M. J. Goodwin, M. J. Aryee, and J. K. Joung. 2014. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature Biotechnology 32(6):569-576.
- Tsai, S. Q., Z. Zheng, N. T. Nguyen, M. Liebers, V. V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A. J. Iafrate, L. P. Le, M. J. Aryee, and J. K. Joung. 2015. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotech- nology 33(2):187-197.
- Wakayama, T., A. C. Perry, M. Zuccotti, K. R. Johnson, and R. Yanagimachi. 1998. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394(6691):369-374.
- Wan, H., Z. He, M. Dong, T. Gu, G. Z. Luo, F. Teng, B. Xia, W. Li, C. Feng, X. Li, T. Li, L. Shuai, R. Fu, L. Wang, X. J. Wang, X. Y. Zhao, and Q. Zhou. 2013. Parthenogenetic haploid embryonic stem cells produce fertile mice. Cell Research 23(11):1330-1333.
- Wang, H., H. Yang, C. S. Shivalila, M. M. Dawlaty, A. W. Cheng, F. Zhang, and R. Jaenisch. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/ Cas-mediated genome engineering. Cell 153(4):910-918.
- Wang, X., Y. Wang, X. Wu, J. Wang, Y. Wang, Z. Qiu, T. Chang, H. Huang, R. J. Lin, and J. K. Yee. 2015. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nature Biotechnology 33(2):175-178.
- Wiedenheft, B., K. Zhou, M. Jinek, S. M. Coyle, W. Ma, and J. A. Doudna. 2009. Structural basis for DNase activity of a conserved protein implicated in CRISPR-mediated genome defense. Structure 17(6):904-912.
- Wiedenheft, B., G. C. Lander, K. Zhou, M. M. Jore, S. J. J. Brouns, J. van der Oost, J. A. Doudna, and E. Nogales. 2011. Structures of the RNA-guided surveillance complex from a bacterial immune system. Nature 477(7365):486-489.
- Wilmut, I., A. E. Schnieke, J. McWhir, A. J. Kind, and K. H. Campbell. 1997. Viable offspring derived from fetal and adult mammalian cells. Nature 385(6619):810-813.
- Wu, Y., H. Zhou, X. Fan, Y. Zhang, M. Zhang, Y. Wang, Z. Xie, M. Bai, Q. Yin, D. Liang, W. Tang, J. Liao, C. Zhou, W. Liu, P. Zhu, H. Guo, H. Pan, C. Wu, H. Shi, L.Wu, F. Tang, and J. Li. 2015. Correction of a genetic disease by CRISPR-Cas9-mediated gene editing in mouse spermatogonial stem cells. Cell Research 25(1):67-79.
- Yang, H., L. Shi, B. A. Wang, D. Liang, C. Zhong, W. Liu, Y. Nie, J. Liu, J. Zhao, X. Gao, D. Li, G. L. Xu, and J. Li. 2012. Generation of genetically modified mice by oocyte injection of androgenetic haploid embryonic stem cells. Cell 149(3):605-617.
- Yang, H., H. Wang, C. S. Shivalila, A. W. Cheng, L. Shi, and R. Jaenisch. 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154(6):1370-1379.
Zeng, H., S. Wen, W. Xu, Z. He, G. Zhai, Y. Liu, Z. Deng, and Y. Sun. 2015. Highly efficient editing of the actinorhodin polyketide chain length factor gene in Streptomyces coelicolor M145 using CRISPR/Cas9-CodA(sm) combined system. Applied Microbiology and Biotechnology 99(24):10575-10585. - Zetsche, B., J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. Van der Oost, A. Regev, E. V. Koonin, and F. Zhang. 2015. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163(3):759-771.
- Zhou, Q., M. Wang, Y. Yuan, X. Wang, R. Fu, H. Wan, M. Xie, M. Liu, X. Guo, Y. Zheng, G. Feng, Q. Shi, X.Y. Zhao, J. Sha, and Q. Zhou. 2016. Complete meiosis from embryonic stem cell-derived germ cells in vitro. Cell Stem Cell 18(3):330-340.
- Zijlstra, J., E. Li, F. Sajjadi, S. Subramani, and R. Jaenisch. 1989. Germ line transmission of a disrupted b2-microglobulin gene produced by homologous recombination in embryonic stem cells. Nature 342(6248):435-438.