L’épilepsie est l’un des troubles chroniques neurologiques les plus courants, avec une prévalence estimée de 0,5 à 1 %. Actuellement, les options de traitement de l’épilepsie reposent principalement sur l’administration d’un traitement symptomatique. La plupart des patients sont en mesure d’atteindre l’absence de crise par les deux premiers essais de médicaments appropriés. Ainsi, les patients qui ne peuvent pas atteindre une réponse satisfaisante après cela sont définis comme pharmacorésistants. Cependant, malgré la disponibilité de plus de 20 médicaments antiépileptiques, environ un tiers des épilepsies restent résistantes aux médicaments. L’hétérogénéité des crises et des épilepsies, la coexistence de comorbidités et le large spectre d’efficacité, d’innocuité et de tolérabilité liés aux antiépileptiques rendent la prise en charge de ces patients réellement difficile. Dans cette revue, nous analysons les problèmes cliniques et pathogéniques les plus pertinents liés à l’épilepsie résistante aux médicaments, puis nous discutons des données actuelles sur l’utilisation des antiépileptiques disponibles et des approches alternatives non pharmacologiques.
Au cours des 20 dernières années, un nombre important de médicaments antiépileptiques, plus précisément définis comme des antiépileptiques, ont été développés et homologués. Cependant, au moins 30 % des personnes atteintes d’épilepsie souffrent d’épilepsie pharmacorésistante, elles restent donc réfractaires aux traitements pharmacologiques courants (1). La Ligue internationale contre l’épilepsie (ILAE) désigne l’épilepsie résistante aux médicaments comme l’échec d’essais adéquats de deux schémas d’antiépileptiques tolérés, choisis et utilisés de manière appropriée, que ce soit en monothérapie ou en association, pour obtenir une absence durable de crises (2). Selon cette définition, une étude observationnelle a analysé la réponse aux antiépileptiques chez des patients hospitalisés atteints d’épilepsie nouvellement diagnostiquée et a montré que les premier et deuxième schémas thérapeutiques étaient efficaces dans 49,5 et 36 %, respectivement. À cet égard, toutes les thérapies avec antiépileptiques après l’échec des deux premiers médicaments ont eu un taux de réussite significativement plus faible (de 12,5 à 22,2 %) (3).
Selon ces données, les chances de contrôler les crises semblent diminuer drastiquement après l’échec du deuxième antiépileptique. Certains cliniciens éviteraient d’essayer d’autres traitements pharmacologiques chez les patients susceptibles de bénéficier d’un traitement chirurgical avec un pourcentage de réussite plus élevé (4). Le concept de pharmacorésistance implique non seulement des crises réfractaires, mais la pathogenèse sous-jacente est responsable de changements structurels et neurobiochimiques qui provoquent également des troubles cognitifs et neuropsychiatriques et un dysfonctionnement psychosocial (5). En raison de l’extrême variabilité des phénotypes des patients atteints d’épilepsie pharmacorésistante et de l’absence d’une définition partagée unique de l’efficacité de l’antiépileptique pour éliminer les crises, il reste difficile de comparer les essais cliniques et de définir des directives de pratique. Cependant, selon l’ILAE, qui a tenté d’en donner une définition opérationnelle, un antiépileptique est jugé efficace s’il existe une période sans crise de 12 mois classiquement ou d’au moins trois fois le plus long intervalle inter-crise avant traitement (2). Le traitement pharmacologique doit être choisi de manière appropriée pour le syndrome épileptique et le type de crises et administré pendant au moins 6 mois à une posologie adéquate (6).
La gamme de doses efficaces optimales et la fréquence d’administration dépendent de la réponse individuelle, des comorbidités en cours et de la tolérance aux médicaments, les effets secondaires indésirables limitant davantage le choix des antiépileptiques (7). Par conséquent, il est clair que les patients atteints de toucher rectal représentent un éventail de tableaux cliniques et neurobiologiques différents plutôt qu’un groupe de patients atteints de la même maladie, nécessitant une approche complexe de plusieurs problèmes sur lesquels nous essaierons de nous concentrer. L’objectif de cette revue est de donner un aperçu des problèmes cliniques et pathogéniques les plus pertinents liés à l’épilepsie pharmacorésistante et d’examiner une approche pratique avec les antiépileptiques, le potentiel de nouveaux antiépileptiques émergents et des thérapies non pharmacologiques alternatives pour les patients atteints d’épilepsie pharmacorésistante.
Le fardeau de l’épilepsie résistante aux médicaments
Malgré la grande variation dans la définition de l’épilepsie pharmacorésistante, une revue épidémiologique systématique récente a rapporté une proportion d’incidence globale de l’épilepsie pharmacorésistante allant de 0,06 à 0,51 % et une prévalence allant de 0,11 à 0,58 %. Parmi les études analysées, l’estimation groupée de la prévalence de l’épilepsie pharmacorésistante était de 0,30 (IC à 95 % : 0,19 à 0,42), ce qui correspond à ce qui a été fréquemment rapporté dans la littérature. La proportion d’incidence regroupée était de 0,15 (IC à 95 % : 0,11 à 0,19) chez les enfants et de 0,34 % (IC à 95 % : 0,06 à 0,62) chez les adultes, avec une incidence globale regroupée de 20 % (IC à 95 % : 0,14 à 0,27) ( 8).
Les causes courantes d’échec du traitement sont une mauvaise observance et une sélection inappropriée de l’antiépileptique en raison d’un diagnostic erroné des crises d’épilepsie. Une relation de confiance avec les patients et leurs familles est indispensable pour assurer une bonne compréhension du problème et une adhésion optimale au schéma thérapeutique prescrit. En revanche, une «fausse pharmacorésistance» doit être écartée chez tout patient présentant des crises difficiles à traiter. Les causes de l’épilepsie pharmacorésistante apparente sont le diagnostic erroné d’événements psychogènes non épileptiques comme des convulsions et l’incapacité d’identifier le bon type d’épilepsie conduisant à une sélection inappropriée de médicaments. D’autres problèmes pouvant conduire à une «fausse pharmacorésistance» sont la prescription d’un dosage inadéquat du médicament et une évaluation incorrecte de la réponse au traitement avec un surdiagnostic des effets secondaires (c’est-à-dire des interactions médicamenteuses entraînant une augmentation des effets secondaires et une mauvaise tolérance) (9).
Comme complication supplémentaire, même les patients correctement étiquetés comme pharmacorésistants peuvent avoir des phases de longue rémission complète (pharmacorésistant) alternant avec une évolution récurrente (pharmacorésistant). Cependant, dans une étude de cohorte d’adultes atteints d’épilepsie pharmacorésistante, parmi les patients avec une période de rémission des crises de 12 mois, le risque de rechute des crises est resté élevé (71,2 % à 5 ans). Par conséquent, il est raisonnable d’être prudent lorsque l’on discute de la probabilité d’une rémission stable (10). Il convient de noter que les personnes atteintes d’épilepsie pharmacorésistante ont un risque de mort subite de 2 à 10 fois supérieur à celui de la population générale. La mort subite et inattendue dans l’épilepsie (MSIE) est définie comme la mort soudaine et inattendue d’une personne atteinte d’épilepsie qui est par ailleurs en bonne santé, dans laquelle aucune autre cause de décès ne peut être trouvée lors d’une autopsie. Le décès survient souvent pendant la nuit, et il peut être témoin ou non, sans qu’il soit nécessaire de prouver une crise convulsive récente (11). Le risque de MSIE est inversement lié au contrôle des crises, en particulier chez les patients présentant un taux plus élevé de crises convulsives et une épilepsie de longue date (12, 13). Les causes de ce décès lié à l’épilepsie ont été largement analysées dans plusieurs études, mais des facteurs prédictifs précis de MSIE font encore défaut. À cet égard, la seule façon de prévenir cette complication chez les patients pharmacorésistants est d’optimiser le contrôle des crises.
Outre la MSIE et d’autres causes de décès prématuré chez les personnes atteintes de toucher rectal (par exemple, accidents liés à des convulsions, néoplasmes cérébraux et maladies neurodégénératives sous-jacentes aux épilepsies symptomatiques), de nombreuses comorbidités différentes peuvent affecter ces patients. Les troubles cognitifs et neuropsychiatriques tels que la dépression et l’anxiété chez les adultes, le déficit de l’attention avec hyperactivité, les troubles du spectre autistique et les problèmes neurocomportementaux chez les enfants sont plus fréquents que dans la population générale (14, 15). Ces comorbidités aggravent les compétences linguistiques et sociales, affectant négativement le fonctionnement psychosocial à long terme. Le spectre des handicaps peut varier considérablement, résultant d’une étiopathologie structurelle, fonctionnelle ou génétique sous-jacente qui a conduit à des convulsions ou à l’effet négatif des thérapies antiépileptiques. Dans de rares cas sélectionnés, une thérapie ciblée sur la cause sous-jacente de l’épilepsie peut améliorer à la fois les crises et les dysfonctionnements cognitifs, comme le régime cétogène pour le syndrome de déficience GLUT1. Dans de nombreuses encéphalopathies épileptiques, l’initiation rapide d’un traitement antiépileptique efficace peut améliorer les symptômes cognitifs et neurocomportementaux (16).
Cependant, ces comorbidités pourraient persister malgré une thérapie antiépileptique adéquate avec absence de crise, peut-être en raison d’une réorganisation synaptique ou d’une neurogenèse altérée, et encore plus représenter un problème de prise en charge complémentaire chez les patients atteints de toucher rectal. En outre, d’autres maladies telles que la migraine, les troubles cardiovasculaires, l’asthme, l’arthrose et le reflux gastro-œsophagien ont une incidence plus élevée chez les personnes atteintes d’épilepsie. Les mécanismes d’association identifiés varient selon les facteurs de risque occasionnels, causaux, résultants, bidirectionnels ou partagés. Par exemple, l’épilepsie peut être causée par des comorbidités telles que des maladies cérébrovasculaires ou un accident vasculaire cérébral périnatal, ou vice versa une pneumonie par aspiration, et une fracture liée à une crise peut être considérée comme une comorbidité résultante de l’épilepsie. Parfois, les conditions partagent les mêmes facteurs de risque ou le même trouble génétique responsable des deux, ou dans d’autres cas, la relation est bidirectionnelle comme dans les troubles du spectre autistique (17).
Facteurs de risque prédictifs
Discerner les patients qui présentent un risque plus élevé de réfractaire aux médicaments pourrait être utile. Plusieurs études ont tenté d’identifier les facteurs prédictifs du développement de l’épilepsie pharmacorésistante, mais elles manquent toutes d’une définition commune unique de la réfractaire aux médicaments et sont affectées par l’hétérogénéité de la population examinée. Diagnostiquer certains types de syndromes épileptiques à l’âge pédiatrique signifie déjà donner une évaluation pronostique de la résistance aux médicaments : le syndrome de Lennox-Gastaut (LGS), l’encéphalopathie épileptique infantile précoce, le syndrome de Dravet (DS) ou l’encéphalite de Rasmussen sont presque pharmacorésistants, peut-être en raison de la schéma neurobiologique sous-jacent à l’épilepsie (7). Plusieurs facteurs cliniques ont été associés à l’épilepsie pharmacorésistante, tels que l’âge au début de l’épilepsie (<1 an), l’étiologie, une neuroimagerie anormale, la coexistence de troubles neuropsychiatriques ou de déficience intellectuelle, des antécédents de convulsions fébriles prolongées ou d’état de mal épileptique et la présence de anomalies EEG spécifiques (18).
L’apparition des crises au cours de la période néonatale est plus susceptible d’être associée à un risque plus élevé de développer un toucher rectal que l’apparition de l’épilepsie plus tard dans la vie. Les épilepsies idiopathiques présentent un risque de pharmacorésistance plus faible que l’épilepsie symptomatique, c’est-à-dire une épilepsie avec des anomalies structurelles sous-jacentes telles qu’une dysplasie corticale, une sclérose temporale mésiale, une sclérose tubéreuse ou des lésions vasculaires. Il est suggéré que les crises focales présentent un risque plus élevé que les crises généralisées (18). Le sexe n’est pas considéré comme un facteur de risque, alors que les antécédents familiaux sont encore controversés selon différentes études (9, 18). Le nombre de crises survenant au cours de l’année précédant le début du traitement, les antécédents d’abus de drogues et les antécédents familiaux d’épilepsie chez les parents au premier degré étaient positivement associés au toucher rectal, selon Chen et ses collègues (18).
Dans l’ensemble, dans une cohorte de patients atteints d’épilepsie nouvellement diagnostiquée de tous types, plus de la moitié des patients sont devenus sans crise pendant le traitement avec un premier antiépileptique, environ 15 % sont devenus sans crise pendant le traitement avec un deuxième ou un troisième médicament, tandis que seulement 3% des cas d’épilepsie ont été contrôlés par un traitement avec deux médicaments. Cela suggère qu’une réponse inadéquate à la thérapie antiépileptique initiale est plus probablement associée au développement d’une épilepsie réfractaire (19). En outre, le temps nécessaire pour atteindre l’absence de crises pourrait également être une valeur pronostique pour les résultats à long terme chez les patients épileptiques. Selon une analyse post-hoc sur des patients traités pour une épilepsie focale, les patients sans crises à 6 mois avaient 90 % de chances de ne plus avoir de crises à 12 mois, alors que ceux qui n’étaient pas sans crises à 6 mois n’avaient que 45 % de chance d’être sans crise à 12 mois, ce qui suggère que la réponse clinique à 6 mois était un excellent prédicteur de la réponse à 12 mois (20).
Pathogénèse
Les mécanismes sous-jacents à l’épilepsie pharmacorésistante ne sont pas complètement connus. La pathogenèse de la résistance aux médicaments est probablement variable et multifactorielle, plusieurs mécanismes agissant ensemble chez un patient donné (6). À cet égard, outre les preuves cliniques, les modèles expérimentaux permettent de mieux caractériser et comprendre les mécanismes putatifs de l’échec de l’antiépileptique. Les mécanismes hypothétiques de la résistance aux médicaments peuvent être entremêlés et comprendre des mécanismes liés à la maladie, des mécanismes liés aux médicaments et des mécanismes génétiques. Selon «l’hypothèse du transporteur», l’expression ou la fonction accrue des protéines d’efflux multidrogues dans le tissu cérébral épileptique humain et dans les modèles animaux de DRE diminue l’efficacité des antiépileptiques, quelle que soit leur cible d’action (21, 22).
La glycoprotéine P dépendante de l’ATP (P-gp) est le produit du gène humain multidrug-resistance-1 (MDR1; ABCB1) et, en raison de sa large spécificité de substrat, elle joue un rôle dans la restriction de l’entrée cérébrale de plusieurs médicaments différents. . Cette protéine est située dans les cellules endothéliales des capillaires cérébraux qui forment la barrière hématoencéphalique (BHE) et agit avec d’autres protéines de résistance multidrogues afin de protéger le cerveau de l’intoxication par des xénobiotiques lipophiles qui autrement traverseraient la barrière hématoencéphalique par diffusion passive (22, 23). Ce mécanisme est à la base de l’échec du traitement de différentes tumeurs cérébrales, d’infections cérébrales et de plusieurs autres troubles cérébraux (22). Plusieurs antiépileptiques présentent des structures chimiques similaires aux substrats de la P-gp, ainsi l’expression accrue de la P-gp et d’autres pompes à efflux peut limiter leur entrée à travers la barrière hématoencéphalique, conférant un phénotype d’épilepsie multirésistant aux médicaments (24). Cependant, les preuves à l’appui de la surexpression de la P-gp dans les tissus cérébraux épileptogènes sont controversées, le rôle d’autres protéines multirésistantes aux médicaments étant encore inconnu (21).
La surexpression des protéines d’efflux de médicaments semble être limitée au foyer épileptique épargnant les tissus normaux adjacents, expliquant l’absence de moins d’effets secondaires neurotoxiques chez les patients réfractaires aux antiépileptiques que chez les patients réactifs (25). En outre, la question de savoir si la surexpression chez les patients DRE est intrinsèque (génétique) ou acquise à la suite de crises incontrôlées ou d’un traitement chronique par l’antiépileptique reste incertaine. Les preuves expérimentales appuient l’hypothèse transportée puisque les non-répondeurs aux antiépileptiques montrent une expression plus élevée de P-gp au niveau de la barrière hématoencéphalique que les répondeurs dans les modèles de rats. De plus, la surexpression de la P-gp est associée à des niveaux cérébraux plus faibles d’antiépileptiques chez les rongeurs, et surtout, l’inhibition de la P-gp par le tariquidar contrecarre la résistance aux antiépileptiques dans un modèle de rat d’épilepsie du lobe temporal (TLE) (11, 22) . Cependant, l’hypothèse du transporteur est encore controversée et d’autres études sont nécessaires pour mieux évaluer la pertinence clinique de la surexpression des transporteurs d’efflux au niveau de la barrière hématoencéphalique.
L ‘«hypothèse pharmacocinétique» suggère que la surexpression des transporteurs d’efflux est localisée dans les organes périphériques, tels que le foie, l’intestin et les reins, diminuant ainsi les taux plasmatiques d’antiépileptiques et la quantité d’antiépileptiques disponible pour traverser la barrière hématoencéphalique. À cet égard, l’expression des transporteurs multidrogues n’est pas nécessairement limitée au cerveau mais peut également se produire dans d’autres organes et tissus. Les études animales ne soutiennent pas cette hypothèse, et les preuves en sa faveur sont assez limitées (11).
Alternativement, «l’hypothèse cible» postule que les altérations induites par l’épilepsie acquise de la structure ou de la fonctionnalité des molécules cibles des antiépileptiques entraînent une réduction de leur réponse au traitement (26). Cette théorie est principalement basée sur des études avec la carbamazépine (CBZ) sur les canaux sodiques voltage-dépendants dans les neurones de l’hippocampe de patients atteints de TLE mésial. Le bloc dépendant de l’utilisation des canaux tension-sodium des cellules granulaires dentées par la CBZ a été complètement perdu chez les patients atteints d’épilepsie résistante à la CBZ, par rapport aux neurones de patients sans TLE mésial. Les mêmes résultats ont été obtenus en bloquant les canaux tension-sodium avec des modèles de rat pilocarpine pour la CBZ et la phénytoïne (PHT) (26, 27), mais pas avec d’autres antiépileptiques tels que la lamotrigine (LTG) et l’acide valproïque (VPA) (27). On ne sait toujours pas si la perte de sensibilité des canaux sodiques chez les patients résistants à la CBZ pourrait être étendue à d’autres antiépileptiques ayant des mécanismes d’action similaires. Conformément à l’hypothèse de la cible, d’autres sites d’action comme les récepteurs GABAa ont été étudiés. Une altération de la sensibilité des récepteurs GABAa a été rapportée dans des modèles animaux de toucher rectal, mais aucune preuve clinique ne l’étaye (9, 28). Par conséquent, même si l’hypothèse cible semble concevable, les preuves disponibles à ce jour sont encore limitées.
De plus, l’épilepsie peut induire des altérations structurelles telles que la neurodégénérescence, la germination axonale, la réorganisation synaptique, la neurogenèse et la gliose, qui sont à la base de «l’hypothèse du réseau neuronal» (29). Ces changements provoquent la formation d’un réseau de neurones anormal qui conduirait à la résistance des antiépileptiques. La sclérose hippocampique soutient cette théorie; ainsi, on pense qu’il joue un rôle causal dans l’apparition de la pharmacorésistance dans le TLE, tandis que la suite d’une résection chirurgicale inverse souvent la résistance (30). Cependant, les altérations du réseau neuronal ne conduisent pas à une réfractaire chez tous les patients épileptiques, ce qui suggère la nécessité d’autres facteurs causaux agissant ensemble (24). Les preuves expérimentales mettent en évidence les conclusions selon lesquelles les dommages de l’hippocampe sont liés à la résistance de l’antiépileptique dans un modèle de rat de DRE.
Au contraire, « l’hypothèse de la gravité intrinsèque » considère la pharmacorésistance comme une propriété inhérente de l’épilepsie liée à la gravité de la maladie (31). Une fréquence élevée des crises est un facteur fiable de pharmacorésistance ; cependant, ce n’est pas le seul prédicteur de la pharmacorésistance. Les modèles animaux présentant une fréquence de crises très élevée sont généralement des non-répondeurs, même si certains des rats épileptiques non-répondeurs peuvent également avoir une faible fréquence de crises. Par conséquent, peu de preuves appuient cette hypothèse. L’« hypothèse génétique » identifie dans les variantes de polymorphismes uniques des gènes la raison de la sensibilité différente à la pharmacorésistance chez les patients atteints d’épilepsie (32). Cette hypothèse est basée sur le concept qu’il existe une variation endogène chez les personnes atteintes d’épilepsie, ce qui réduit les chances de contrôler les crises avec les antiépileptiques. Malheureusement, il n’y a pas encore d’associations génétiques généralement acceptées pour soutenir le modèle des mécanismes de résistance aux médicaments indépendants du syndrome entraînés par la variation génétique, car jusqu’à présent, les études disponibles sont de taille limitée, impliquant différents groupes de patients et uniquement des variantes de polymorphismes uniques sélectionnés ( par exemple, SCN1A, ABCB1) (33).
La neuroinflammation et le dysfonctionnement de la barrière hématoencéphalique peuvent jouer un rôle important en tant que mécanismes potentiels dans la promotion et le maintien de l’activité épileptique (34–36). Une perméabilité améliorée de la barrière hématoencéphalique est présente dans les modèles expérimentaux et les conditions cliniques. Un dysfonctionnement de la barrière hématoencéphalique est généralement associé à une réponse neuro-inflammatoire concomitante dans les mêmes zones tissulaires. Dans toutes ces conditions, il y a une induction de la P-gp dans les vaisseaux cérébraux et les astrocytes. La neuroinflammation et le dysfonctionnement de la barrière hématoencéphalique sont donc les caractéristiques d’une zone épileptogène dans diverses formes d’épilepsie pharmacorésistante et dans des modèles animaux d’épilepsies acquises (11). À cet égard, des études précliniques sur des modèles expérimentaux de crises aiguës et d’épilepsie chronique ont montré que la neuroinflammation dans les zones cérébrales d’apparition et de généralisation des crises joue un rôle central dans l’hyperexcitabilité neuronale sous-jacente à la génération des crises. La microglie et les astrocytes sont impliqués de manière cruciale à la fois dans l’induction et la perpétuation de la réponse inflammatoire aux lésions ou crises épileptogènes ; les autres contributeurs sont les neurones, les composants cellulaires de la barrière hématoencéphalique et les leucocytes (37). De plus, des molécules et des voies inflammatoires spécifiques ont été identifiées pour influencer les résultats dans divers modèles expérimentaux d’épilepsie (38). De plus en plus de preuves démontrent que la neuroinflammation, les changements de perméabilité de la barrière hématoencéphalique, le dysfonctionnement astrocytaire conduisent à l’épileptogenèse, à la progression de l’épilepsie et à la résistance à l’antiépileptique (11, 37, 38).
Les encéphalopathies à médiation par anticorps sont des maladies inflammatoires du cerveau de plus en plus reconnues comme cause de convulsions et d’états de mal épileptiques, réfractaires aux MSA classiques mais pouvant bénéficier d’une immunothérapie. Un diagnostic précoce de ces affections conduit à un traitement adapté avec des agents immunomodulateurs et, dans la plupart des cas, le traitement symptomatique par les antiépileptiques peut être interrompu après la phase aiguë. Le risque ultérieur de développer une épilepsie chronique est relativement faible (10 à 15 %) et peut varier en fonction de l’antigène cible et de l’immunothérapie rapide. L’épilepsie chronique après encéphalopathies à médiation par anticorps est généralement caractérisée par des crises résistantes aux médicaments qui peuvent résulter d’un processus inflammatoire en cours qui persiste au-delà de la phase aiguë ou sous forme de séquelles dues à des modifications irréversibles altérant les réseaux neuronaux et persistant après la résolution du processus inflammatoire (34, 39).
Thérapie pharmacologique dans l’épilepsie résistante aux médicaments
Monothérapie vs polythérapie rationnelle à la nouvelle ère des antiépileptiques
Malgré les avancées récentes dans le domaine de l’épilepsie et l’introduction de nouveaux antiépileptiques au cours des 20 dernières années, la prise en charge des épilepsie pharmacorésistante reste complexe et laisse de nombreuses questions sans réponse. Le traitement pharmacologique est le pilier de la prise en charge de l’épilepsie réfractaire, et dans cette classe de patients, la polythérapie doit être soigneusement évaluée, en tenant compte du rapport bénéfice/risque en termes d’efficacité et de tolérance, ainsi que de l’observance du patient. Les questions les plus importantes posées par les médecins concernant le choix des antiépileptiques chez les patients atteints d’épilepsie pharmacorésistante sont “comment puis-je sélectionner les patients atteints d’épilepsie pharmacorésistante candidats à la polythérapie, et comment puis-je mettre en place le schéma thérapeutique optimal pour eux ?”. L’absence de lignes directrices fondées sur des données probantes pouvant guider le médecin dans le choix des schémas thérapeutiques les plus efficaces et les plus appropriés complique la prise en charge des patients atteints d’épilepsie pharmacorésistante. Cependant, nous discutons plus en détail des recommandations théoriques qui peuvent guider le clinicien vers la «polythérapie rationnelle» pour le toucher rectal. Ce processus nécessite des essais systématiques d’antiépileptiques après l’échec d’au moins deux régimes médicamenteux correctement essayés.
Les antiépileptiques de première génération étaient limités en nombre, en mécanismes d’action, en pharmacocinétique (inducteurs ou inhibiteurs puissants) et en profil de tolérance en raison d’un taux élevé d’événements indésirables (EI). Un seul essai contrôlé randomisé (ECR) comparant la CBZ en monothérapie à une association de CBZ et d’APV en tant que schéma thérapeutique initial chez des patients souffrant de crises tonico-cloniques généralisées et/ou partielles non traitées a été mené : les résultats étaient en faveur de la thérapie combinée même si ce n’est pas le cas. statistiquement significatif, mais cette combinaison a montré des interactions médicamenteuses pharmacocinétiques pertinentes (40). Avant d’être acceptés sur le marché, les nouveaux antiépileptiques ont fait l’objet d’ECR rigoureux chez des patients prenant de un à trois antiépileptiques, montrant la supériorité des nouveaux antiépileptiques par rapport au placebo en tant que thérapie complémentaire. Les preuves montrent de meilleurs profils pharmacologiques, y compris une pharmacocinétique linéaire, moins de potentiel d’interactions médicamenteuses et différents mécanismes d’action qui peuvent être combinés dans la thérapie d’association. Après de nombreuses années d’utilisation en polythérapie, de nouveaux antiépileptiques ont été étudiés dans des ECR comparatifs en monothérapie. Certains d’entre eux sont désormais également indiqués comme médicaments de première intention pour les crises focales ou certains types d’épilepsie (41–43).
L’objectif de la polythérapie chez les patients pharmacorésistants est d’identifier des combinaisons d’antiépileptiques qui maximisent l’efficacité et minimisent les effets secondaires (40). Il existe des études cliniques humaines limitées sur les meilleures combinaisons d’antiépileptiques; par conséquent, le choix d’un deuxième ou d’un troisième médicament dans une polythérapie rationnelle devrait également tenir compte des études animales et des considérations empiriques (44). Des modèles animaux expérimentaux ont permis d’évaluer les interactions pharmacodynamiques en thérapie combinée qui sont principalement liées aux mécanismes d’action des antiépileptiques, par analyse isobolographique ou mesure directe de l’indice thérapeutique. Les effets souhaitables de la polythérapie devraient être un effet supra-additif anti-épileptique (effet de synergie) et éventuellement un antagonisme neurotoxique ou un effet infra-additif neurotoxique (45, 46).
Une incidence plus élevée d’EI et une efficacité plus faible ont été détectées dans des essais cliniques qui comparaient des combinaisons de médicaments ayant des effets bloquant les canaux sodiques, par rapport à une thérapie combinée d’un inhibiteur des canaux sodiques et d’un médicament avec un mécanisme d’action différent comme un médicament GABAergique (47– 49). D’où l’hypothèse d’interactions synergiques des antiépileptiques pilotées par des mécanismes d’action, selon laquelle le choix d’une combinaison d’antiépileptiques basée sur différents mécanismes d’action peut augmenter l’efficacité du traitement. Dans une vaste étude basée sur la population de patients souffrant de crises focales, il est apparu que les régimes d’antiépileptiques avec différents mécanismes d’action avaient de meilleurs résultats à la fois en termes de durée de traitement et de risque d’hospitalisation et d’admission aux urgences (50).
La combinaison avec la meilleure preuve de synergie dans les études humaines est VPA avec LTG. L’interaction synergique entre ces antiépileptiques a été rapportée dans plusieurs études, qui ont démontré un taux de réponse majeur avec l’utilisation de LTG comme traitement d’appoint avec l’APV par rapport à l’ajout de LTG à la CBZ ou à la PHT (51). L’association du VPA à l’éthosuximide (ETX) était considérée comme plus efficace que le VPA ou l’ETX en monothérapie chez les enfants présentant des crises d’absence difficiles à traiter (52). Les autres schémas thérapeutiques combinés étudiés dans une cohorte de patients présentant des crises focales sont le LTG-LEV (53) et le lacosamide (LCM)-LEV (54, 55). L’effet synergique de l’association de ces antiépileptiques peut résulter de l’association de différents mécanismes d’action. En effet, le LCM améliore l’inactivation lente des canaux sodiques voltage-dépendants, tandis que le LEV module la libération des neurotransmetteurs synaptiques en se liant à la protéine de la vésicule synaptique SV2A dans le cerveau (53, 54). Des schémas associant LTG-TPM et VPA-LEV chez l’adulte pourraient être utiles, mais avec un faible niveau de preuve (56, 57). Un bon niveau de preuve existe pour l’utilisation du Stiripentol (STP) comme traitement d’appoint en association avec le clobazam (CLB) et l’APV chez les enfants et les adolescents atteints de SD. Le STP a été approuvé comme traitement d’appoint pour le DS en Europe en 2007, suivi au Japon (2012), au Canada (2012) et aux États-Unis (2018) (58).
Deux essais contrôlés réalisés en 2000 (59) et 2002 (60) chez de jeunes patients atteints de SD qui recevaient déjà un traitement VPA et CLB ont montré un taux de réponse significativement plus élevé chez ceux traités avec STP que le placebo. Ces résultats ont été confirmés par des études observationnelles rétrospectives et prospectives à long terme ultérieures en termes de contrôle des crises, de réduction des crises prolongées, du nombre d’épisodes d’état de mal épileptique et d’hospitalisations. Les effets anti-épileptiques du STP observés dans la SD peuvent résulter de deux mécanismes différents. Une pharmacocinétique, qui fournit l’augmentation des métabolites actifs du CLB médiée par le STP, et une concernant le mécanisme d’action du STP, est une amélioration de la transmission acidergique gamma-aminobutyrique par les récepteurs GABA post-synaptiques dans un site d’action différent des benzodiazépines (58 ). Enfin, d’autres nouveaux antiépileptiques tels que l’acétate d’eslicarbazépine, la gabapentine et le zonisamide se sont avérés efficaces en tant que médicaments supplémentaires dans le traitement de l’épilepsie pharmacorésistante (44). Cependant, même avec l’intérêt croissant pour les nouveaux antiépileptiques et leur utilisation recommandée dans le DRE, aucun d’entre eux n’a prouvé sa supériorité sur les antiépileptiques conventionnels dans des études de comparaison directe en termes d’efficacité (61).
Les autres enjeux concernant l’utilisation de la polythérapie rationnelle sont les différents profils pharmacocinétiques et EI et les conséquences des interactions médicamenteuses. Comme mentionné précédemment, les antiépileptiques plus anciens ont de nombreuses interactions avec d’autres médicaments (antiépileptiques ou autres), principalement médiées par leur effet sur le cytochrome P450 en tant qu’inhibiteurs ou inducteurs enzymatiques. Par exemple, lorsque l’on considère l’association entre le VPA et le LTG, il est important de considérer que le VPA agit comme un puissant inhibiteur métabolique enzymatique ; ainsi, il peut réduire la clairance du LTG, augmentant son niveau hématique et, par conséquent, la probabilité d’hypersensibilité induite par le LTG ou de développement de tremblements (62). Dans ce concours, le médecin doit lentement titrer LTG, en commençant par des doses plus faibles de la dernière (44). La CBZ, le phénobarbital (PB) et le PHT sont plutôt des inducteurs-enzymes qui peuvent réduire les niveaux d’anticoagulants, de contraceptifs oraux ou d’immunosuppresseurs. De plus, ces antiépileptiques plus anciens présentent un spectre d’EI allant de l’hépatotoxicité et de l’encéphalopathie associées à l’APV à la suppression de la moelle osseuse induite par la CBZ, la PHT et la PB (63). A l’inverse, les nouveaux antiépileptiques ont moins d’interactions pharmacocinétiques (la plupart sont des inducteurs ou inhibiteurs enzymatiques faibles) et de meilleurs profils de tolérance ; par conséquent, ils sont des candidats optimaux pour la thérapie combinée (64).
À cet égard, le LEV a le moins d’interactions pharmacologiques et, avec le TPM et le zonisamide, il est largement utilisé en polythérapie. Certains nouveaux antiépileptiques ont des contre-indications spécifiques, comme le LCM, le rufinamide et la retigabine chez les patients atteints du syndrome du QT long, ainsi que le TPM, le zonisamide et le LEV chez les patients présentant des comorbidités neuropsychiatriques en raison du risque accru d’anxiété, de dépression et de psychose (44) . Il est intéressant de souligner que le recours à la polythérapie n’implique pas nécessairement une augmentation des EI. Selon certaines études, le risque d’EI était similaire entre les patients en monothérapie et en polythérapie, et les patients appartenant au second groupe étaient même capables de tolérer une charge médicamenteuse totale plus élevée (TDL, rapport de la dose journalière prescrite définie par l’OMS) par rapport à ceux en monothérapie. Les auteurs ont suggéré que la survenue des EI en polythérapie n’était pas strictement liée au nombre de médicaments mais plutôt au type et à la posologie des antiépileptiques choisis et à la sensibilité individuelle (65, 66) (voir tableau 1).
Tableau 1. Dans ce tableau, les principales combinaisons d’antiépileptiques dans plusieurs études humaines sont présentées.
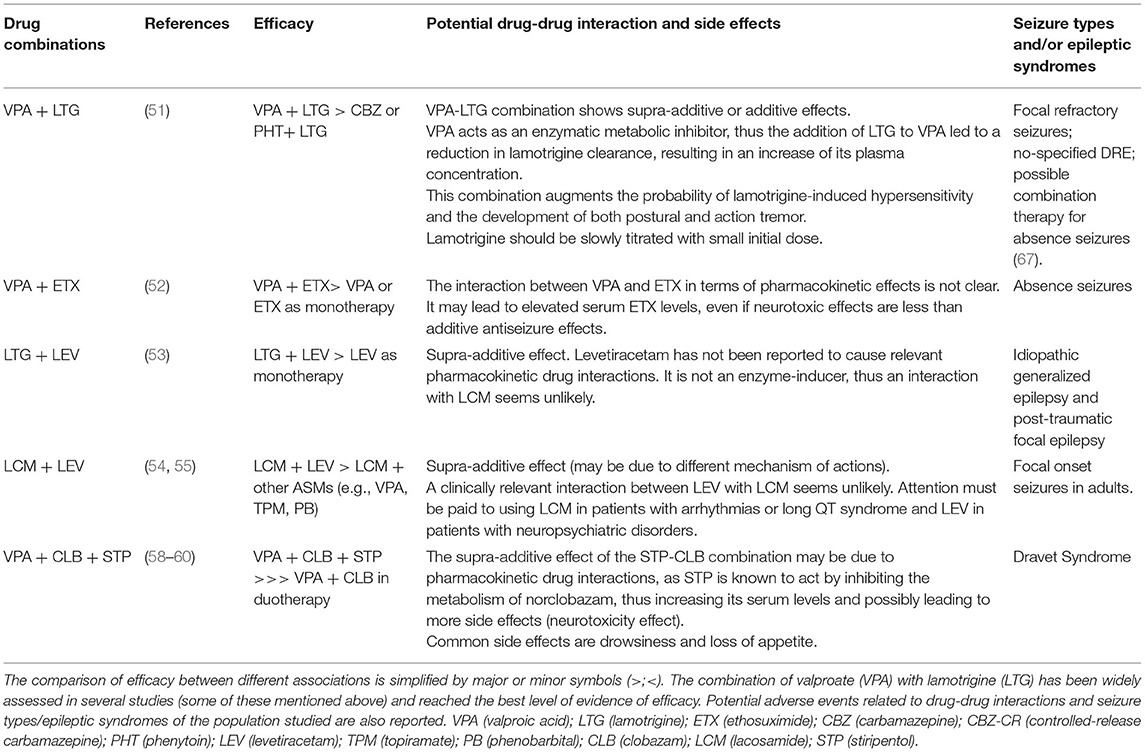 Par conséquent, le choix d’un régime optimal d’antiépileptiques pour un patient pharmacorésistant dans une polythérapie rationnelle doit tenir compte de nombreuses variables non seulement concernant les aspects pharmacodynamiques et pharmacocinétiques des antiépileptiques, mais également des facteurs liés au patient tels que l’âge, l’observance, les comorbidités et les médicaments concomitants. Enfin, le syndrome d’épilepsie et les types de crises devraient guider le médecin dans le choix des meilleurs antiépileptiques dans la prise en charge du toucher rectal.
Par conséquent, le choix d’un régime optimal d’antiépileptiques pour un patient pharmacorésistant dans une polythérapie rationnelle doit tenir compte de nombreuses variables non seulement concernant les aspects pharmacodynamiques et pharmacocinétiques des antiépileptiques, mais également des facteurs liés au patient tels que l’âge, l’observance, les comorbidités et les médicaments concomitants. Enfin, le syndrome d’épilepsie et les types de crises devraient guider le médecin dans le choix des meilleurs antiépileptiques dans la prise en charge du toucher rectal.
Médicaments anticonvulsivants dans les syndromes épileptiques réfractaires
Les syndromes épileptiques idiopathiques comprennent une grande variété d’épilepsies focales et généralisées, affectant principalement les enfants ou les adolescents, qui ont en commun une origine génétique connue ou présumée et un schéma électroclinique reconnaissable. La majorité de ces patients présentent généralement une rémission spontanée ou parviennent à ne plus avoir de crises avec un traitement, mais un sous-ensemble d’entre eux (20 à 30 %) peut présenter des crises persistantes malgré un traitement antiépileptique de première et de deuxième ligne. Les options de traitement du syndrome d’épilepsie-absence de l’enfant (ECA) pharmacorésistant qui ne répond pas à l’ETX, au VPA ou au LTG en monothérapie en tant que MSA de premier choix, pourraient bénéficier de l’association du VPA et du LTG (67). Comme indiqué précédemment, cette combinaison a montré une interaction synergique et peut être considérablement plus efficace utilisée en duothérapie plutôt que l’une ou l’autre séparément. L’association du VPA et de l’ETX dans l’ECA s’est avérée efficace chez les enfants réfractaires au VPA ou à l’ETX en monothérapie (52), bien qu’aucune étude plus récente n’ait comparé l’association VPA-ETX au VPA-LTG.
D’autres antiépileptiques pouvant être essayés en cas d’échec de ces options comprennent le CLB, le zonisamide et le TPM ; notamment, le PHT, le CBZ et les barbituriques sont inefficaces et contre-indiqués dans les crises d’absence (67, 68). En outre, les encéphalopathies développementales et épileptiques bien définies telles que DS, LGS et l’encéphalopathie épileptique précoce de la petite enfance sont plutôt constitutivement pharmacorésistantes et nécessitent presque toujours une thérapie combinée. Dans le contexte des encéphalopathies développementales et épileptiques, le SD représente un prototype de syndrome épileptique pharmacorésistant (69). Il s’agit d’une maladie génétique principalement associée à des mutations de perte de fonction dans SCN1A, un gène codant pour les canaux sodiques voltage-dépendants, avec la perte d’action qui en résulte dans les interneurones ergiques de l’acide gamma-aminobutyrique (GABA).
Comme mentionné précédemment, le STP a d’abord été approuvé comme traitement d’appoint dans le DS en association avec le VPA et le CLB, qui sont les médicaments de première intention dans la prise en charge pharmacologique de ces patients. L’utilisation du TPM pourrait être considérée comme une thérapie de deuxième intention comme alternative au STP dans le traitement d’appoint avec le CLB et le VPA (70). En outre, des essais cliniques émergents ont démontré l’efficacité dans le contrôle des crises de nouvelles thérapies pharmacologiques pour le DS, discutées plus en détail ci-dessous dans le texte : le cannabidiol (CBD) et la fenfluramine (FFA). Le CBD a atteint des preuves d’efficacité de classe I en tant qu’antiépileptique dans le DS en plus d’autres médicaments et pourrait être utilisé comme traitement de deuxième ligne avec ou sans STP. Il existe des preuves cliniques prometteuses pour l’utilisation de FFA à faible dose dans le DS, mais l’efficacité et l’innocuité doivent encore être confirmées (58).
Le LGS est l’une des encéphalopathies épileptiques pédiatriques les plus graves, représentant 1 à 10 % de toutes les épilepsies infantiles et un pic d’incidence entre la 3e et la 5e année. Il a une étiologie relativement hétérogène (génétique, structurelle, métabolique ou inconnue), un schéma spécifique de plusieurs types de crises (toniques, atoniques, crises de chute), des anomalies EEG et une réfraction typique au traitement avec seulement de courtes périodes de rémission (71). Les ECR de monothérapie et la comparaison directe des antiépileptiques d’appoint font actuellement défaut pour ces patients. L’APV est toujours considérée comme la thérapie de première intention pour le traitement du LGS nouvellement diagnostiqué. La LTG est souvent considérée comme le meilleur choix en cas d’échec de l’APV, avec la plus grande efficacité contre les crises d’épilepsie, alors qu’elle est contre-indiquée dans les crises myocloniques en raison de son effet potentiel d’aggravation. Le rufinamide, le TPM et le CLB se sont également avérés efficaces en thérapie combinée avec un bon niveau de preuve. Le zonisamide, le LEV, le pérampanel (PER) et le FFA pourraient avoir une efficacité possible dans ce syndrome, bien que le niveau de preuve ne soit pas encore cohérent (71). En revanche, le CBD a obtenu une bonne classe de preuves d’efficacité dans le LGS comme dans le DS ; par conséquent, son utilisation doit être encouragée chaque fois que d’autres traitements ont échoué (72, 73).
Une approche pratique pour la prise en charge des patients atteints d’épilepsie résistante aux médicaments
Généralement, les patients atteints d’épilepsie nouvellement diagnostiqués sont initialement traités par une monothérapie puisque les preuves montrent que 50 % des patients atteints d’épilepsie non traitée obtiennent un contrôle prolongé des crises (> 12 mois) avec un premier schéma thérapeutique (74). Selon plusieurs experts, la polythérapie ne devrait être envisagée qu’après l’échec d’au moins deux ou trois monothérapies (75–77). Néanmoins, des preuves récentes suggèrent de choisir la duothérapie comme alternative à la deuxième monothérapie médicamenteuse après l’échec de la monothérapie initiale, obtenant ainsi une rémission des crises de 15 à 20 % avec environ 60 % des patients qui peuvent parvenir à une libération des crises après le deuxième essai médicamenteux (41, 78).
Les patients qui n’ont pas répondu au deuxième essai médicamenteux, en monothérapie ou en duothérapie, répondent aux critères de l’ILAE pour la définition du toucher rectal et doivent être référés à des centres dédiés à l’épilepsie (2). Cependant, ils doivent être réévalués (c’est-à-dire avec une vidéo-EEG prolongée, une neuroimagerie) pour éventuellement poser le bon diagnostic de type d’épilepsie ou exclure les causes de pseudo-pharmacorésistance mentionnées ci-dessus dans cette revue. Les patients qui peuvent bénéficier d’un traitement chirurgical comme ceux causés par des lésions épileptogènes focales ont une probabilité plus élevée de rémission des crises (environ 60 à 80 %) s’ils sont rapidement orientés vers des centres spécialisés plutôt que de poursuivre les essais ultérieurs de médicaments.
Dans le cas contraire, si la lésion ne peut être complètement réséquée chirurgicalement sans séquelles neurologiques conséquentes, des essais systématiques d’une deuxième duothérapie ou d’une trithérapie peuvent être nécessaires, avec un pourcentage de rémission plus faible (41, 78). Ainsi, la sélection de candidats potentiels à la polythérapie représente une étape cruciale et conduit à des implications pratiques pertinentes afin d’augmenter les possibilités de contrôle des crises. Néanmoins, l’ajout d’un quatrième médicament doit être plutôt évité, car l’utilisation de plus de trois médicaments augmente la probabilité d’EI, par rapport à une amélioration limitée du contrôle des crises (79). En cas d’échec d’un 5e ou 6e essai médicamenteux avec duo-triple ou quadruple thérapie, d’autres thérapies alternatives doivent être évaluées. La stimulation du nerf vague (VNS) ou le régime cétogène peuvent être activement poursuivis. Si le patient a déjà été traité avec des antiépileptiques précédents, il est crucial d’évaluer soigneusement les doses d’administration, l’efficacité et le spectre des EI des médicaments précédemment administrés.
Médicaments anticonvulsivants émergents pour vaincre l’épilepsie résistante aux médicaments
Malgré le développement d’un grand nombre d’antiépileptiques au cours des dernières décennies, environ 30 % des patients épileptiques sont réfractaires au traitement médical et ont un besoin urgent de nouvelles options de traitement pour contrôler avec succès les crises et améliorer leur qualité de vie (2). En raison de la genèse multifactorielle de l’épilepsie pharmacorésistante et de la difficulté à comprendre comment différents mécanismes pourraient interagir chez le même patient ou groupe de patients, la tâche de surmonter la résistance aux médicaments avec de nouveaux traitements pharmacologiques reste encore difficile.
Les MSA récemment approuvées comme traitement d’appoint chez les patients présentant des crises focales, avec ou sans généralisation secondaire, et des crises tonico-cloniques généralisées primaires sont la PER et le brivaracétam (BRV). Le PER est un antagoniste des récepteurs α-amino-3-hydroxyl-5-méthyl-4-isoxazole-propionate (AMPA) non compétitif, premier de sa catégorie, et il agit comme un antiépileptique potentiellement à large spectre. L’efficacité de la PER chez les patients atteints d’épilepsie focale résistante aux médicaments a été évaluée dans 3 essais cliniques de phase III randomisés en double aveugle contrôlés par placebo (ECR 304, 305, 306) et dans un essai d’extension en ouvert (étude 307) (80–84) . L’utilisation approuvée du PER pour les crises tonico-cloniques généralisées primaires (PGTC) était basée sur un essai randomisé en double aveugle contrôlé par placebo (étude 332) qui fournit des preuves de classe I ILAE de la réduction de la fréquence des crises dans l’épilepsie généralisée idiopathique réfractaire (étude 332) (85, 86).
Le BRV est actuellement approuvé pour le traitement d’appoint chez les patients souffrant de crises d’épilepsie focales. Semblable au lévétiracétam (LEV), le BRV agit en se liant aux vésicules SV2A avec une haute affinité et un profil pharmacocinétique linéaire. Chez les adultes atteints d’épilepsie focale réfractaire aux médicaments, la BRV d’appoint s’est avérée efficace pour réduire la fréquence des crises et assez bien tolérée. D’autres études sont nécessaires pour tirer des conclusions définitives sur son efficacité chez les participants non naïfs au LEV et évaluer son profil d’innocuité à long terme (87). De plus en plus de preuves soutiennent également sa prescription aux patients pédiatriques grâce à son profil d’efficacité et de tolérabilité (88).
Parmi les nouvelles molécules étudiées pour le toucher rectal, le CBD et le FFA ont démontré de bonnes preuves d’efficacité dans les essais cliniques sur le toucher rectal. Dans l’un des premiers essais cliniques, le CBD – le composé non psychoactif dérivé du cannabis – a été approuvé en tant que solution orale de CBD purifié en thérapie combinée avec le CLB chez les patients DS et LGS à partir de l’âge de 2 ans (72). Le CBD a une structure chimique et un mécanisme d’action distinctifs par rapport aux autres nouveaux antiépileptiques et représente le premier de cette nouvelle classe de médicaments. À des concentrations cliniquement significatives, il ne montre pas d’effets psychoactifs, mais il agit sur de multiples cibles en tant que molécule anti-épileptique, y compris l’antagonisme du récepteur couplé aux protéines G 55 (GPR55), la désensibilisation du potentiel de récepteur transitoire de type vanilloïde 1 (TRPV1) canaux et modulation allostérique positive des récepteurs GABAa (72, 89).
Un premier essai de phase II, randomisé, en double aveugle et contrôlé par placebo sur le CBD a été réalisé sur des enfants et des adolescents atteints de SD (90, 91) ; puis deux essais de phase III, randomisés, en double aveugle et contrôlés par placebo ont démontré l’efficacité et l’innocuité du CBD en tant que médicament d’appoint chez les patients atteints de LGS, en particulier pour le contrôle des crises d’épilepsie (92, 93). Une étude d’extension en ouvert a confirmé ces données (94). Aucune corrélation dose-réponse (10 contre 20 mg/kg/jour) n’a été trouvée dans une méta-analyse ultérieure (95), mais des preuves récentes ont montré que le CBD d’appoint à la dose de 10 ou 20 mg/kg/jour entraînait des résultats similaires. réductions de la fréquence des crises convulsives pour les doses de 10 et 20 mg/kg/jour, avec un meilleur profil d’innocuité et de tolérabilité pour la dose de 10 mg/kg/jour (96). Les EI courants liés au CBD étaient les vomissements, la fatigue, la pyrexie, la diminution de l’appétit, la somnolence, la léthargie et la diarrhée (72). Il reste à déterminer si l’un des effets thérapeutiques et indésirables du CBD pourrait être lié à l’association avec le CLB en tant que médicament concomitant. En effet, des preuves suggèrent que le CBD affecte de manière significative les niveaux de CLB/N-desméthylclobazam, tout au long de l’effet sur le cytochrome p450 en tant qu’enzyme inhibitrice (97).
Cependant, même l’effet synergique de probabilité, des données robustes indiquent que le CBD est à la fois efficace avec ou sans association avec le CLB (98-100). D’autre part, l’utilisation des AGL comme antiépileptique est apparue de manière inhabituelle puisqu’elle a d’abord été approuvée comme médicament amaigrissant, puis retirée en 1997 en raison d’effets cardiaques (hypertrophie valvulaire et hypertension pulmonaire) (101). Le FFA est un dérivé de l’amphétamine et exerce son effet anticonvulsivant en perturbant le stockage de la sérotonine dans les vésicules, en inhibant sa recapture à partir de la synapse et en modulant positivement le récepteur sigma 1. De plus, son métabolite norfenfluramine montre une forte affinité pour les récepteurs de la sérotonine dans le cerveau (en particulier sur 5HT2C et 1D, 5HT2A non clair) (102). Ainsi, les FFA ont continué à être utilisés, comme cela a été rapporté dans de nombreuses séries de cas, chez des enfants atteints de différents types d’épilepsie, parmi lesquels des patients pharmacorésistants (58). Un groupe de neurologues pour enfants en Belgique a continué à utiliser les FFA chez les enfants atteints de SD à des doses plus faibles, montrant des résultats encourageants (103). L’efficacité de l’utilisation des FFA dans le contrôle des crises a été analysée dans plusieurs études portant sur des patients pédiatriques atteints de SD et de LGS (104-107).
Dans une étude prospective en ouvert impliquant des patients atteints de DS traités par AGL à la dose moyenne de 0,35 (0,16–0,69) mg/kg/jour pendant une période médiane de 1,5 an, il en est résulté une réduction médiane des crises de 75 % ( 104). Deux essais cliniques randomisés multicentriques, en double aveugle, contrôlés par placebo, incluant des enfants atteints de SD traités avec des schémas thérapeutiques contenant des FFA incluant le STP (avec une dose variable de 0,2 à 0,7 mg/kg/jour, maximum 30 mg/jour), ont montré une réponse plus importante taux en terme de réduction des crises convulsives dans le groupe traité par FFA par rapport au placebo, sans effet secondaire cardiaque observé (105, 106). L’absence d’EI cardiaques résultait de doses plus faibles utilisées comme antiépileptique (20 à 30 mg/jour maximum) par rapport aux doses de médicaments amaigrissants (jusqu’à 60 mg/jour) (108). Les EI non cardiovasculaires les plus courants étaient l’anorexie, la diarrhée, la rhinopharyngite, la léthargie, la somnolence et la pyrexie. Les mêmes résultats d’efficacité, de sécurité et de tolérabilité, même s’ils sont moins cohérents en termes de nombre d’essais cliniques réalisés, ont été obtenus pour l’utilisation des FFA dans le LGS à différents schémas posologiques (de 0,1 à 0,8 mg/kg/jour). Des essais contrôlés randomisés sont toujours en cours ; ainsi dans un futur proche, nous pourrons confirmer ces résultats (102, 107).
Outre le CBD et le FFA, il existe également des études prometteuses sur l’utilisation du cénobamate et du padsevonil, largement discutées par Loscher et al. (11). Selon «l’hypothèse cible» mentionnée ci-dessus, l’une des approches actuelles dans la recherche de nouvelles molécules efficaces pour le DRE est le développement d’antiépileptiques plus efficaces par la découverte révisée de médicaments basés sur la cible. Dans ce scénario, il a été conçu le padsevonil, un composé avec un mécanisme d’action similaire au LEV et au brivaracétam, mais avec une affinité plus élevée sur les protéines SV2 présynaptiques et un effet plus large sur différents sous-types SV2. Actuellement, le padsevonil fait l’objet d’un essai de phase III chez des patients souffrant de crises focales multirésistantes (109). Le cénobamate a été étudié avec le même raisonnement, et c’est un nouvel antiépileptique récemment approuvé pour le traitement des crises partielles chez les adultes, qui agit en améliorant les courants inhibiteurs par la modulation des récepteurs GABAa et en diminuant le courant sodique excitateur (110).
Une autre approche intéressante à l’ère de la génomique est la « médecine de précision ». L’avènement des technologies génomiques permet désormais de mieux caractériser le fond génétique de l’épilepsie, et change lentement la façon de classer les syndromes épileptiques. Différents modèles de mutations génétiques pourraient sous-tendre le même syndrome épileptique et être responsables de différentes réponses aux médicaments, et des mutations du même gène peuvent entraîner différents phénotypes (11). La liste des gènes porteurs de variants pathogènes rares s’allonge rapidement. Ces découvertes ont conduit à des stratégies de traitement rationnelles, y compris une meilleure sélection des antiépileptique parmi les médicaments existants ou réorientés qui n’étaient pas utilisés auparavant pour l’épilepsie.
En effet, dans certains cas, il est possible de corriger des défauts métaboliques spécifiques (ex. le régime cétogène pour le déficit en GLUT1, ou la pyridoxine pour les épilepsies pyridoxino-dépendantes) (111), d’éviter les antiépileptiques qui peuvent potentiellement aggraver le défaut pathogène (ex. l’administration de médicaments bloquant les canaux sodiques dans le DS lié à SCN1A), contraster le défaut fonctionnel causé par une mutation génique en utilisant des antiépileptiques existants (par exemple, en utilisant des inhibiteurs des canaux sodiques dans les variants pathogéniques SCN8A ou SCN2A avec effet de gain de fonction), ou en utilisant des médicaments déjà disponibles sur le marché pour d’autres indications (par exemple, la mémantine utilisée pour traiter l’encéphalopathie épileptique causée par la mutation GRIN2A des récepteurs NMDA du glutamate) (112). Néanmoins, les deux seuls exemples validés de cette médecine de précision sont l’utilisation de l’évérolimus dans l’épilepsie focale associée au complexe de la sclérose tubéreuse et l’utilisation du CBD et des FFA dans le DS, alors que la plupart des traitements spécifiques aux gènes intéressants rapportés ne sont basés que sur des rapports de cas. ou études de courte durée (113).
Alternatives au traitement pharmacologique de l’épilepsie résistante aux médicaments
L’alternative la meilleure et potentiellement résolutive aux antiépileptiques pour les patients atteints d’épilepsie réfractaire est le traitement chirurgical, chaque fois que cela est possible. Dans certains cas, retarder la chirurgie peut aggraver les chances post-chirurgicales d’éviter les crises ; par conséquent, il est crucial d’identifier rapidement les patients qui sont des candidats potentiels à l’intervention, comme indiqué ci-dessus. Les lésions focales couramment réséquées dans l’épilepsie focale comprennent la sclérose hippocampique et la dysplasie corticale focale, avec un plus grand succès chez les patients présentant des lésions IRM concordantes avec les crises cliniques (114, 115). Cependant, chez un nombre important de patients, la zone épileptogène ne peut être identifiée ou traitée chirurgicalement en raison de sa localisation au sein du tissu cérébral fonctionnel. Pour ce groupe de patients, la neurostimulation devient un traitement alternatif ou complémentaire aux médicaments de plus en plus accepté (116). De nos jours, plusieurs modalités de neurostimulation sont disponibles pour le toucher rectal, soit invasives, nécessitant une intervention chirurgicale pour implanter le dispositif, soit non invasives, sans implantation permanente de dispositif nécessaire. Certains dispositifs fournissent une stimulation continue (boucle ouverte), tandis que d’autres délivrent une stimulation basée sur l’activité cérébrale détectée (boucle fermée).
Parmi les méthodes invasives, l’approche de neurostimulation la plus étudiée et la plus établie est la VNS, suivie de la stimulation cérébrale profonde (DBS), de la neurostimulation réactive plus récente (RNS) et de la stimulation corticale chronique sous le seuil (CSCS). Les alternatives non invasives sont la stimulation transcutanée du nerf vague (tVNS), la stimulation du nerf trijumeau (TNS), la stimulation magnétique transcrânienne (TMS) et la stimulation transcrânienne à courant continu (tDCS) (117, 118). Malheureusement, malgré le nombre croissant d’appareils de neurostimulation, les connaissances sur les mécanismes sous-jacents sont relativement limitées et il existe un manque de lignes directrices ou de consensus général dans les centres d’épilepsie sur le moment et la manière d’utiliser ces traitements. Le VNS a été approuvé aux États-Unis par la Food and Drug Administration (FDA) comme traitement d’appoint pour le toucher rectal en 1997, mais son utilisation a également été étudiée dans d’autres domaines de la médecine (par exemple, la dépression, l’insuffisance cardiaque, les accidents vasculaires cérébraux et les acouphènes) ( 119). Bien qu’initialement approuvé pour les crises partielles chez les personnes âgées de plus de 12 ans, le VNS est maintenant utilisé chez les adultes et les enfants atteints de toucher rectal non éligibles à la chirurgie de résection, souffrant de crises focales ou généralisées (120).
Cette approche consiste en une stimulation électrique périodique délivrée au nerf vague fournie par un générateur d’impulsions programmable, généralement implanté en sous-cutané sous la clavicule gauche et connecté à un fil conducteur enroulé autour du nerf vague en aval du nerf laryngé récurrent. La stimulation du nerf vague gauche est préférée car on pense qu’elle a moins d’effets cardiaques probables (par exemple, bradycardie) plutôt que le nerf droit qui innerve directement le nœud sino-auriculaire (119). Le mécanisme d’action exact du VNS sur le contrôle des crises n’est pas encore entièrement compris. Il implique peut-être d’abord le noyau tractus solitarius, qui se projette par conséquent dans d’autres régions cruciales telles que le locus coeruleus, les noyaux du raphé, le thalamus, l’hypothalamus et le circuit limbique. Par conséquent, la modulation des projections noradrénergiques et sérotoninergiques semble pertinente, comme en témoigne l’augmentation des niveaux de sérotonine ou de son précurseur chez les patients traités par VNS. Concordant avec l’effet anti-épileptique, d’autres changements d’acides aminés dans le SNC rapportés après VNS sont des niveaux accrus du neurotransmetteur inhibiteur GABA et des niveaux réduits d’aspartate d’acide aminé excitateur (121).
Une fonction altérée du système limbique, du système d’activation réticulaire et des structures corticales (c’est-à-dire le cortex orbitofrontal, les lobes temporaux) montrée par la neuroimagerie fonctionnelle, et la preuve encore plus forte de l’effet désynchronisant sur l’enregistrement EEG du cuir chevelu du VNS sont supposées contribuer à l’anticonvulsion effet de ce traitement (119). Le VNS traditionnel inclut la stimulation de base (boucle ouverte) et le mode aimant (à la demande). La stimulation de base est le paradigme de fonctionnement principal dans lequel l’appareil effectue continuellement des cycles avec une stimulation intermittente active 24 h par jour avec des périodes d’activation et de désactivation (par exemple, 30 s allumés et 5 min éteints). La stimulation en mode aimant permet au patient ou au personnel soignant de délivrer une stimulation complémentaire lors d’une crise, déclenchée en passant un aimant sur le générateur d’impulsions (122). Le VNS réactif est une approche moderne basée sur l’auto-stimulation en boucle fermée, qui délivre automatiquement une stimulation déclenchée par la tachycardie critique, un marqueur de l’apparition des crises survenant dans > 80 % des crises généralisées et focales (119, 122). Généralement, il est recommandé de commencer par une stimulation de base 2 semaines après l’implantation du dispositif.
Dans le réglage de l’appareil, les paramètres de stimulation à moduler comprennent la largeur d’impulsion (à partir de 250–500 μs), la fréquence des impulsions (à partir de 20 à 30 Hz) et la durée des temps de marche/arrêt (généralement 30 s marche et 5 min off). Le courant de sortie est initialement réglé sur 0,25 mA et progressivement augmenté jusqu’à des niveaux thérapeutiques (1,25–2,0 mA), en fonction de la réponse clinique et de la tolérance du patient. Les preuves issues des principaux ECR réalisés sur des patients résistants aux médicaments traités par VNS ont montré un taux de répondeurs (réduction d’au moins 50 % de la fréquence des crises) de 26 à 40 % (123). Le paradigme de stimulation élevée a entraîné une réduction significative du taux de crises par rapport au VNS à basse fréquence (risque relatif IC à 95 % allant de 1,13 à 2,64) (124). Un seul ECR a été mené chez des enfants et n’a montré aucune différence statistiquement significative dans la réduction de la fréquence des crises entre les stimulations élevées et faibles (125). VNS est bien toléré, sans différences significatives en termes d’EI entre les deux paradigmes de stimulation. Les principaux EI signalés sont l’enrouement, la toux, la dyspnée, la douleur, la paresthésie, les nausées et les maux de tête (124). Des preuves de haute qualité provenant d’ECR se sont concentrées sur les épilepsies focales chez les patients adultes atteints de toucher rectal, et cependant des études observationnelles suggèrent que le VNS pourrait également être efficace dans les épilepsies généralisées (119).
La DBS est un traitement invasif non pharmacologique approuvé par la FDA en 2018 pour le toucher rectal chez l’adulte (>18 ans) lorsque la résection chirurgicale est contre-indiquée. Une stimulation électrique prédéterminée (en boucle ouverte) des structures cérébrales profondes telles que le noyau antérieur du thalamus (ANT), l’hippocampe (HC), le noyau centromédian du thalamus (CMT), le cervelet et le globus pallidus, est délivrée tout au long de l’implantation électrodes reliées à un générateur d’impulsions. Bien que le mécanisme ne soit pas complètement compris, on pense que le DBS perturbe les réseaux responsables de la propagation des crises dans lesquelles l’activité thalamique est impliquée, entraînant une réduction des décharges interictales (118). Dans l’essai SANTE (Stimulation of the Anterior Nucleus of the Thalamus for Epilepsy), 110 patients adultes atteints d’épilepsie localisée ont été recrutés et ont montré une réduction médiane du taux de crises de 40 % après la phase en aveugle de 3 mois et de 69 % après 5 mois. années de suivi (126, 127).
Selon une étude récente, il a été démontré que la stimulation du noyau antérieur du thalamus (ANT) et de l’hippocampe (HC) diminue la fréquence des crises réfractaires, la moitié de tous les patients inscrits dans des études cliniques expérimentant une réduction des crises de 46 à 90 % pour ANT-DBS et 48 à 95 % avec HC-DBS (128). Au contraire, moins de preuves d’efficacité sont disponibles pour la stimulation d’autres cibles, et aucun ECR n’a impliqué de patients pédiatriques (118, 128). Même s’ils sont généralement bien tolérés, les EI les plus fréquents signalés dans l’essai SANTE étaient des douleurs au site d’implantation et des paresthésies (126). Un taux plus élevé de dépression et de troubles de la mémoire a été détecté dans le groupe actif par rapport à la stimulation factice, même s’il n’a pas été confirmé lors de l’étude de suivi de 5 ans (127). Il convient de noter que certains types et syndromes de crises semblent plus sensibles à la stimulation de cibles spécifiques, telles que la stimulation HC pour les épilepsies temporales et la stimulation CMT pour les LGS et les crises généralisées (129, 130).
Une autre approche de neurostimulation pour le toucher rectal est le RNS, qui est basé sur un dispositif en boucle fermée capable de détecter des schémas spécifiques d’activité épileptogène et de fournir une stimulation focale pour interrompre l’activité épileptique. Il se compose d’un générateur d’impulsions implanté sous le cuir chevelu, d’une sonde de profondeur placée à l’aide d’un logiciel stéréotaxique dans la zone d’apparition critique ou d’une sonde sous-durale implantée à travers un trou de fraise et positionnée sur la zone corticale souhaitée, et d’un programmateur externe grâce auquel il est possible de moduler les paramètres de détection et de stimulation en fonction des caractéristiques du patient (131).Les principales indications du RNS sont le traitement des patients adultes atteints de toucher rectal focal, qui ont des crises focales avec pas plus de deux foyers épileptogènes et qui ont trois crises invalidantes ou plus par mois. En plus de la réduction de la fréquence des crises, le RNS permet une électrocorticographie à long terme, utile pour suivre les détections de crises au fil du temps ou pour déterminer la latéralité exacte du début des crises (118, 131). Un ECR a montré une réduction médiane des crises de 53 % chez 256 patients adultes 2 ans après l’implantation (132). Comme démontré dans d’autres modalités de neurostimulation (par exemple, DBS), les pourcentages de réduction des crises et le taux de réponse ont continué à s’améliorer au fil du temps dans les études de suivi (133-135). Aucun EI grave n’a été signalé, mais ceux typiques d’autres dispositifs de neurostimulation, alors que l’amélioration de la qualité de vie et des domaines cognitifs a été enregistrée à long terme (134). Au contraire, il n’existe aucune preuve adéquate de l’utilisation du SRN à l’âge pédiatrique.
Semblable à la VNS, la stimulation corticale sous-seuil chronique (CSCS) cible la zone focale corticale ictale mais avec une stimulation sous-seuil continue (boucle ouverte) (136). Cependant, peu de données sur les données cliniques sont aujourd’hui disponibles et uniquement basées sur des études rétrospectives. En plus des modalités de neurostimulation invasives décrites ci-dessus, TMS, tDCS, tVNS et TNS émergent récemment comme des approches alternatives non invasives. Dans la TMS, un flux magnétique externe appliqué sur le cuir chevelu génère des courants intracrâniens, qui peuvent exciter des potentiels d’action et moduler des circuits corticaux spécifiques, réduisant ainsi la probabilité de récidive des crises. Même s’il est sûr (l’EI le plus signalé est la migraine après stimulation) et utilisé dans le traitement de plusieurs affections telles que la dépression, la migraine, la douleur ou les troubles du mouvement, les preuves d’efficacité chez les patients atteints d’épilepsie pharmacorésistante sont de faible qualité en raison soit d’un nombre relativement faible et hétérogène de patients inscrits dans les études et résultats non uniques en termes de réduction des crises (137, 138).
Quant à la TMS, l’utilisation de la tDCS a été validée pour de nombreuses maladies neurologiques et psychiatriques (par exemple, Parkinson, douleur, dépression, fonction motrice et cognition) et l’épilepsie également. Le tDCS fournit un courant continu aux régions corticales ciblées grâce à l’application d’électrodes du cuir chevelu (anode et cathode), qui peuvent diminuer l’excitabilité corticale (cathodique), supprimer les crises et interférer avec les décharges épileptiformes observées sur l’EEG (139). Selon différentes études, il existe des preuves de qualité modérée à très faible que la tDCS répétée est efficace chez les patients résistants aux médicaments, en particulier avec l’épilepsie du lobe temporal mésial, la sclérose de l’hippocampe et le LGS (117, 139, 140). Le VNS transcutané (tVNS) et la stimulation du nerf trijumeau (TNS) ont été étudiés dans de petites études. Bien que bien toléré, il existe des preuves de faible qualité de l’efficacité du traitement DRE (117).
En résumé, les données actuelles disponibles confirment l’efficacité et l’innocuité des VNS, DBS, RNS et tDCS pour le toucher rectal chez les patients adultes, principalement atteints d’épilepsie focale. D’autres approches sont généralement bien tolérées, mais ne sont pas encore étayées par des preuves adéquates. Il convient de noter que la prudence s’impose lors de l’application de VNS chez les patients souffrant d’apnée ou de SCP chez ceux souffrant de dépression ou de troubles cognitifs en raison d’une possible aggravation de ces symptômes (117). Parmi les traitements non pharmacologiques du toucher rectal, il y a le régime cétogène. Ce régime alimentaire strict a généralement été réservé à un groupe spécifique d’enfants atteints de toucher rectal et se caractérise par des niveaux élevés de graisses et faibles en glucides, qui imitent le jeûne (141, 142). Cependant, il n’est généralement pas utilisé à long terme en raison de préoccupations concernant les effets sur la croissance et la santé globale. En raison d’un manque d’ECR et du petit nombre de patients inscrits dans les études examinées, les preuves de l’utilisation du régime cétogène pour le toucher rectal sont de qualité faible à très faible ; des recherches supplémentaires sont donc nécessaires, en particulier pour les adultes (143). Néanmoins, s’il est bien toléré, le régime cétogène peut rester une option valable pour les patients résistants aux médicaments.
Conclusions
La prise en charge des patients réfractaires aux médicaments reste assurément un grand défi pour les médecins. L’hétérogénéité des manifestations cliniques, tant en termes de sémiologie que d’évolution des crises, avec des périodes de rémission et de rechute variables et souvent imprévisibles chez les patients atteints de toucher rectal, rend difficile la comparaison des études cliniques et la définition de lignes directrices. Les théories pathogéniques connues à ce jour, basées principalement sur des études précliniques, ne donnent pas une explication unique et intégrée de la résistance aux médicaments, mais seulement de certains mécanismes qui peuvent la sous-tendre et pourraient devenir la cible de nouvelles molécules développées par la recherche. Bien qu’il ne résolve pas le problème et qu’il soit accablé par des effets secondaires inévitables et des interactions médicamenteuses chez les patients subissant une polythérapie, le traitement pharmacologique reste la première et principale approche pour parvenir à un contrôle à long terme des crises. Après une classification diagnostique correcte et la sélection des patients candidats à la chirurgie de résection, il est obligatoire d’avoir une connaissance adéquate des médicaments, avec leur profil pharmacodynamique et pharmacocinétique, les effets indésirables éventuels et les associations pharmacologiques les plus efficaces, afin de choisir le médicament adéquat. régimes adaptés au patient.
Dans la nouvelle ère de l’antiépileptiques de deuxième, troisième et dernière génération, la polythérapie rationnelle a acquis plus de pertinence, grâce au développement de médicaments qui ont des mécanismes d’action différents et potentiellement synergiques, ainsi que de meilleurs profils d’innocuité et d’efficacité par rapport aux antiépileptiques de première génération. Par conséquent, avant de considérer un patient complètement insensible au traitement médicamenteux, il est nécessaire de réévaluer le diagnostic d’épilepsie lui-même et, lorsqu’il est disponible, le patrimoine génétique (comme on le voit, dans certains cas, même la génétique peut conduire à choisir un type de médicament plutôt qu’un autre) et examinez de manière critique les médicaments administrés précédemment pour essayer des schémas thérapeutiques ultérieurs plus adéquats. Des approches non pharmacologiques alternatives telles que la stimulation électrique et la thérapie diététique sont également prometteuses, mais elles ne sont pas résolutives à long terme d’après les preuves dont nous disposons jusqu’à présent.
Conflit d’intérêt
Les auteurs déclarent que la recherche a été menée en l’absence de toute relation commerciale ou financière pouvant être interprétée comme un conflit d’intérêts potentiel.
Références
- Picot MC, Baldy-Moulinier M, Daures JP, Dujols P, Crespel A. The prevalence of epilepsy and pharmacoresistant epilepsy in adults: a population-based study in a Western European country. Epilepsia. (2008) 49:1230–8. doi: 10.1111/j.1528-1167.2008.01579.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia. (2010) 51:1069–77. doi: 10.1111/j.1528-1167.2009.02397.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Brodie MJ, Barry SJE, Bamagous G, Norrie JD, Kwan P. Patterns of treatment response in newly diagnosed epilepsy. Neurology. (2012) 78:1548–54. doi: 10.1212/WNL.0b013e3182563b19 PubMed Abstract | CrossRef Full Text | Google Scholar
- Berg AT. Understanding the delay before epilepsy surgery: who develops intractable focal epilepsy and when? CNS Spectr. (2004) 9:136–44. doi: 10.1017/S109285290000849X PubMed Abstract | CrossRef Full Text | Google Scholar.
- Kwan P, Brodie MJ. Refractory epilepsy: a progressive, intractable but preventable condition? Seizure. (2002) 11:77–84. doi: 10.1053/seiz.2002.0593 PubMed Abstract | CrossRef Full Text | Google Scholar
- Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med. (2011) 365:919–26. doi: 10.1056/NEJMra1004418 CrossRef Full Text | Google Scholar
- Dalic L, Cook MJ. Managing drug-resistant epilepsy: challenges and solutions. Neuropsychiatr Dis Treat. (2016) 12:2605–16. doi: 10.2147/NDT.S84852 PubMed Abstract | CrossRef Full Text | Google Scholar
- Kalilani L, Sun X, Pelgrims B, Noack-Rink M, Villanueva V. The epidemiology of drug-resistant epilepsy: a systematic review and meta-analysis. Epilepsia. (2018) 59:2179–93. doi: 10.1111/epi.14596 PubMed Abstract | CrossRef Full Text | Google Scholar
- Inizio Pati S, Alexopoulos AV. Pharmacoresistant epilepsy: from pathogenesis to current and emerging therapies. Cleve Clin J Med. (2010) 77:457–67 doi: 10.3949/ccjm.77a.09061. PubMed Abstract | CrossRef Full Text | Google Scholar
- Callaghan B, Schlesinger M, Rodemer W, Pollard J, Hesdorffer D, Allen Hauser W, et al. Remission and relapse in a drug-resistant epilepsy population followed prospectively. Epilepsia. (2011) 52:619–26. doi: 10.1111/j.1528-1167.2010.02929.x PubMed Abstract | CrossRef Full Text | Google Scholar.
- Löscher W, Potschka H, Sisodiya SM, Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. (2020) 72:606–38. doi: 10.1124/pr.120.019539 PubMed Abstract | CrossRef Full Text | Google Scholar.
- Nei M, Bagla R. Seizure-related injury and death. Curr Neurol Neurosci Rep. (2007) 7:335–41. doi: 10.1007/s11910-007-0051-1 CrossRef Full Text | Google Scholar.
- Verrotti A, D’Alonzo R, Rinaldi VE, Casciato S, D’Aniello A, Di Gennaro G. Childhood absence epilepsy and benign epilepsy with centro-temporal spikes: a narrative review analysis. World J Pediatr. (2017) 13:106–11. doi: 10.1007/s12519-017-0006-9 PubMed Abstract | CrossRef Full Text | Google Scholar
- Matricardi S, Farello G, Operto FF, Coppola G, Verrotti A. What are the challenges with the pharmacological management of epilepsy in patients with Attention Deficit Hyperactivity Disorder (ADHD)? Exp Opin Pharmacother. (2020) 21:737–9. doi: 10.1080/14656566.2020.1732351 PubMed Abstract | CrossRef Full Text | Google Scholar
- Coppola G, Operto FF, Matricardi S, Verrotti A. Monitoring and managing depression in adolescents with epilepsy: current perspectives. Neuropsychiatr Dis Treat. (2019) 15:2773–80. doi: 10.2147/NDT.S192714 PubMed Abstract | CrossRef Full Text | Google Scholar
- Nickels KC, Zaccariello MJ, Hamiwka LD, Wirrell EC. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol. (2016) 12:465–76. doi: 10.1038/nrneurol.2016.98 PubMed Abstract | CrossRef Full Text | Google Scholar
- Keezer MR, Sisodiya SM, Sander JW. Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. (2016) 15:106–15. doi: 10.1016/S1474-4422(15)00225-2 PubMed Abstract | CrossRef Full Text | Google Scholar
- Chen Z, Brodie MJ, Liew D, Kwan P. Treatment outcomes in patients with newly diagnosed epilepsy treated with established and new antiepileptic drugs: a 30-year longitudinal cohort study. JAMA Neurol. (2018) 75:279–86. doi: 10.1001/jamaneurol.2017.3949 PubMed Abstract | CrossRef Full Text | Google Scholar
- Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. (2000) 342:314–9. doi: 10.1056/NEJM200002033420503 CrossRef Full Text | Google Scholar
- Schmidt D. How reliable is early treatment response in predicting long-term seizure outcome? Epilepsy Behav. (2007) 10:588–94. doi: 10.1016/j.yebeh.2007.02.011 PubMed Abstract | CrossRef Full Text | Google Scholar
- Löscher W, Potschka H. Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci. (2005) 6:591–602. doi: 10.1038/nrn1728 PubMed Abstract | CrossRef Full Text | Google Scholar
- Tang F, Hartz AMS, Bauer B. Drug-resistant epilepsy: multiple hypotheses, few answers. Front Neurol. (2017) 8:301. doi: 10.3389/fneur.2017.00301 PubMed Abstract | CrossRef Full Text | Google Scholar
- König J, Müller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. (2013) 65:944–66. doi: 10.1124/pr.113.007518 PubMed Abstract | CrossRef Full Text | Google Scholar
- Kwan P, Brodie MJ. Potential role of drug transporters in the pathogenesis of medically intractable epilepsy. Epilepsia. (2005) 46: 224–35. doi: 10.1111/j.0013-9580.2005.31904.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Sisodiya SM, Lin WR, Harding BN, Squier MV, Thom M. Drug resistance in epilepsy: expression of drug resistance proteins in common causes of refractory epilepsy. Brain. (2002) 125:22–31. doi: 10.1093/brain/awf002 PubMed Abstract | CrossRef Full Text | Google Scholar
- Remy S, Beck H. Molecular and cellular mechanisms of pharmacoresistance in epilepsy. Brain. (2006) 129:18–35. doi: 10.1093/brain/awh682 PubMed Abstract | CrossRef Full Text | Google Scholar
- Remy S, Urban BW, Elger CE, Beck H. Anticonvulsant pharmacology of voltage-gated Na+ channels in hippocampal neurons of control and chronically epileptic rats. Eur J Neurosci. (2003) 17:2648–58. doi: 10.1046/j.1460-9568.2003.02710.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Bethmann K, Fritschy JM, Brandt C, Löscher W. Antiepileptic drug resistant rats differ from drug responsive rats in GABA a receptor subunit expression in a model of temporal lobe epilepsy. Neurobiol Dis. (2008) 31:169–87. doi: 10.1016/j.nbd.2008.01.005 PubMed Abstract | CrossRef Full Text | Google Scholar
- Fang M, Xi ZQ, Wu Y, Wang XF. A new hypothesis of drug refractory epilepsy: neural network hypothesis. Med Hypotheses. (2011) 76:871–6. doi: 10.1016/j.mehy.2011.02.039 PubMed Abstract | CrossRef Full Text | Google Scholar
- Schmidt D, Löscher W. Drug resistance in epilepsy: putative neurobiologic and clinical mechanisms. Epilepsia. (2005) 46:858–77. doi: 10.1111/j.1528-1167.2005.54904.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Rogawski MA. The intrinsic severity hypothesis of pharmacoresistance to antiepileptic drugs. Epilepsia. (2013) 2:33–40. doi: 10.1111/epi.12182 PubMed Abstract | CrossRef Full Text | Google Scholar
- Balestrini S, Sisodiya SM. Pharmacogenomics in epilepsy. Neurosci Lett. (2018) 667:27–39. doi: 10.1016/j.neulet.2017.01.014 CrossRef Full Text | Google Scholar
- Löscher W, Klotz U, Zimprich F, Schmidt D. The clinical impact of pharmacogenetics on the treatment of epilepsy. Epilepsia. (2009) 50:1–23. doi: 10.1111/j.1528-1167.2008.01716.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Granata T, Fusco L, Matricardi S, Tozzo A, Janigro D, Nabbout R. Inflammation in pediatric epilepsies: update on clinical features and treatment options. Epilepsy Behav. (2021) 107959. doi: 10.1016/j.yebeh.2021.107959 PubMed Abstract | CrossRef Full Text | Google Scholar
- Marchi N, Granata T, Janigro D. Inflammatory pathways of seizure disorders. Trends Neurosci. (2014) 37:55–65. doi: 10.1016/j.tins.2013.11.002 CrossRef Full Text | Google Scholar
- Matricardi S, Farello G, Savasta S, Verrotti A. Understanding childhood neuroimmune diseases of the central nervous system. Front Pediatr. (2019) 7:511. doi: 10.3389/fped.2019.00511 PubMed Abstract | CrossRef Full Text | Google Scholar
- Terrone G, Salamone A, Vezzani A. Inflammation and epilepsy: preclinical findings and potential clinical translation. Curr Pharm Des. (2017) 23:5569–76. doi: 10.2174/1381612823666170926113754 PubMed Abstract | CrossRef Full Text | Google Scholar
- Aronica E, Bauer S, Bozzi Y, Caleo M, Dingledine R, Gorter JA, et al. Neuroinflammatory targets and treatments for epilepsy validated in experimental models. Epilepsia. (2017) 58:27–38. doi: 10.1111/epi.13783 PubMed Abstract | CrossRef Full Text | Google Scholar
- Iorio R, Assenza G, Tombini M, Colicchio G, Della Marca G, Benvenga A, et al. The detection of neural autoantibodies in patients with antiepileptic-drug-resistant epilepsy predicts response to immunotherapy. Eur J Neurol. (2015) 22:70–8. doi: 10.1111/ene.12529 PubMed Abstract | CrossRef Full Text | Google Scholar
- Deckers CL, Hekster YA, Keyser A, Van Lier HJ, Meinardi H, Renier WO. Monotherapy versus polytherapy for epilepsy: a multicenter double-blind randomized study. Epilepsia. (2001) 42:1387–94. doi: 10.1046/j.1528-1157.2001.30800.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Park KM, Kim SE, Lee BI. Antiepileptic drug therapy in patients with drug-resistant epilepsy. J Epilepsy Res. (2019) 9:14–26. doi: 10.14581/jer.19002 PubMed Abstract | CrossRef Full Text | Google Scholar
- Wilmshurst JM, Gaillard WD, Vinayan KP, Tsuchida TN, Plouin P, Van Bogaert P, et al. Summary of recommendations for the management of infantile seizures: task force report for the ILAE commission of pediatrics. Epilepsia. (2015) 56:1185–97. doi: 10.1111/epi.13057 PubMed Abstract | CrossRef Full Text | Google Scholar
- Glauser T, Ben-Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kälviäinen R, et al. ILAE subcommission on AED guidelines: updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. (2013) 54:551–63. doi: 10.1111/epi.12074 PubMed Abstract | CrossRef Full Text | Google Scholar
- Verrotti A, Tambucci R, Di Francesco L, Pavone P, Iapadre G, Altobelli E, et al. The role of polytherapy in the management of epilepsy: suggestions for rational antiepileptic drug selection. Expert Rev Neurother. (2020) 20:167–73. doi: 10.1080/14737175.2020.1707668 PubMed Abstract | CrossRef Full Text | Google Scholar
- Czuczwar SJ, Borowicz KK. Polytherapy in epilepsy: the experimental evidence. Epilepsy Res. (2002) 52:15–23. doi: 10.1016/S0920-1211(02)00181-X CrossRef Full Text | Google Scholar
- Lasoń W, Dudra-Jastrzebska M, Rejdak K, Czuczwar SJ. Basic mechanisms of antiepileptic drugs and their pharmacokinetic/pharmacodynamic interactions: an update. Pharmacol Rep. (2011) 63:271–92. doi: 10.1016/S1734-1140(11)70497-2 PubMed Abstract | CrossRef Full Text | Google Scholar
- Besag FM, Berry DJ, Pool F, Newbery JE, Subel B. Carbamazepine toxicity with lamotrigine: pharmacokinetic or pharmacodynamic interaction? Epilepsia. (1998) 39:183–7. doi: 10.1111/j.1528-1157.1998.tb01356.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Barcs G, Walker EB, Elger CE, Scaramelli A, Stefan H, Sturm Y, et al. Oxcarbazepine placebo-controlled, dose-ranging trial in refractory partial epilepsy. Epilepsia. (2000) 41:1597–607. doi: 10.1111/j.1499-1654.2000.001597.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Sake JK, Hebert D, Isojärvi J, Doty P, De Backer M, Davies K, et al. A pooled analysis of lacosamide clinical trial data grouped by mechanism of action of concomitant antiepileptic drugs. CNS Drugs. (2010) 24:1055–68. doi: 10.2165/11587550-000000000-00000 PubMed Abstract | CrossRef Full Text | Google Scholar
- Margolis JM, Chu BC, Wang ZJ, Copher R, Cavazos JE. Effectiveness of antiepileptic drug combination therapy for partial-onset seizures based on mechanisms of action. JAMA Neurol. (2014) 71:985–93. doi: 10.1001/jamaneurol.2014.808 PubMed Abstract | CrossRef Full Text | Google Scholar
- Brodie MJ, Yuen AW. Lamotrigine substitution study: evidence for synergism with sodium valproate? 105 study group. Epilepsy Res. (1997) 26:423–32. doi: 10.1016/S0920-1211(96)01007-8 PubMed Abstract | CrossRef Full Text | Google Scholar
- Rowan AJ, Meijer JW, De Beer-Pawlikowski N, Van Der Geest P, Meinardi H. Valproate-ethosuximide combination therapy for absence seizures. Arch Neurol. (1983) 40:797–802. doi: 10.1001/archneur.1983.04050120047006 PubMed Abstract | CrossRef Full Text | Google Scholar
- Kinirons P, McCarthy M, Doherty CP, Delanty N. Predicting drug-resistant patients who respond to add-on therapy with levetiracetam. Seizure. (2006) 15:387–92. doi: 10.1016/j.seizure.2006.05.001 PubMed Abstract | CrossRef Full Text | Google Scholar
- Brigo F, Ausserer H, Tezzon F, Nardone R. When one plus one makes three: the quest for rational antiepileptic polytherapy with supraadditive anticonvulsant efficacy. Epilepsy Behav. (2013) 27:439–42. doi: 10.1016/j.yebeh.2013.03.010 PubMed Abstract | CrossRef Full Text | Google Scholar
- Chung S, Ben-Menachem E, Sperling MR, Rosenfeld W, Fountain NB, Benbadis S, et al. Examining the clinical utility of lacosamide: pooled analyses of three phase II/III clinical trials. CNS Drugs. (2010) 24:1041–54. doi: 10.2165/11586830-000000000-00000 PubMed Abstract | CrossRef Full Text | Google Scholar
- Stephen IJ, Sills GJ, Brodie MJ. Lamotrigine and topiramate may be useful combination. Lancet. (1998) 351:958–59. doi: 10.1016/S0140-6736(05)60613-7 CrossRef Full Text | Google Scholar
- Brodie MJ. Pharmacological treatment of drug-resistant epilepsy in adults: a practical guide. Curr Neurol Neurosci Rep. (2016) 16:82. doi: 10.1007/s11910-016-0678-x PubMed Abstract | CrossRef Full Text | Google Scholar
- Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: treatment options and management of prolonged seizures. Epilepsia. (2019) 60:39–48. doi: 10.1111/epi.16334 PubMed Abstract | CrossRef Full Text | Google Scholar
- Chiron C, Marchand MC, Tran A, Rey E, D’Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. (2000) 356:1638–42. doi: 10.1016/S0140-6736(00)03157-3 PubMed Abstract | CrossRef Full Text | Google Scholar
- Guerrini R, Tonnelier S, D’Athis P, Rey E, Vincent J, Pons G, et al. Stiripentol in severe myoclonic epilepsy in infancy (SMEI): a placebo-controlled Italian trial. Epilepsia. (2002) 43:155. doi: 10.1111/j.1528-1167.2002.tb06320.x CrossRef Full Text | Google Scholar
- Lee SK. Old versus new: why do we need new antiepileptic drugs? J Epilepsy Res. (2014) 4:39–44. doi: 10.14581/jer.14010 PubMed Abstract | CrossRef Full Text | Google Scholar
- Zaccara G, Franciotta D, Perucca E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia. (2007) 48:1223–44. doi: 10.1111/j.1528-1167.2007.01041.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Aliyu H, Ayo JO, Ambali SF, Kawu MU, Aluwong T, Dzenda T, et al. Heamatobiochemical alterations induced by carbamazepine and phenytoin: mini review. Biochem Pharmacol. (2016) 5:219. doi: 10.4172/2167-0501.1000219 CrossRef Full Text | Google Scholar
- Kwan P, Brodie MJ. Combination therapy in epilepsy: when and what to use. Drugs. (2006) 66:1817–29. doi: 10.2165/00003495-200666140-00004 CrossRef Full Text | Google Scholar
- Canevini MP, De Sarro G, Galimberti CA, Gatti G, Licchetta L, Malerba A, et al. Relationship between adverse effects of antiepileptic drugs, number of coprescribed drugs, and drug load in a large cohort of consecutive patients with drug-refractory epilepsy. Epilepsia. (2010) 51:797–804. doi: 10.1111/j.1528-1167.2010.02520.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Lammers MW, Hekster YA, Keyser A, Meinardi H, Renier WO, van LierH. Monotherapy or polytherapy for epilepsy revisited: a quantitative assessment. Epilepsia. (1995) 36:440–6. doi: 10.1111/j.1528-1157.1995.tb00484.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Kessler SK, McGinnis E. A practical guide to treatment of childhood absence epilepsy. Paediatr Drugs. (2019) 21:15–24. doi: 10.1007/s40272-019-00325-x PubMed Abstract | CrossRef Full Text | Google Scholar
- Franzoni E, Matricardi S, Di Pisa V, Capovilla G, Romeo A, Tozzi E, et al. Refractory absence seizures: an Italian multicenter retrospective study. Eur J Paediatr Neurol. (2015) 19:660–4. doi: 10.1016/j.ejpn.2015.07.008 PubMed Abstract | CrossRef Full Text | Google Scholar
- Brigo F, Striano P, Balagura G, Belcastro V. Emerging drugs for the treatment of Dravet syndrome. Exp Opin Emerg Drugs. (2018) 23:261–269. doi: 10.1080/14728214.2018.1552937 PubMed Abstract | CrossRef Full Text | Google Scholar
- Wirrell EC, Laux L, Donner E, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American consensus panel. Pediatr Neurol. (2017) 68:18–34. doi: 10.1016/j.pediatrneurol.2017.01.025 PubMed Abstract | CrossRef Full Text | Google Scholar
- Verrotti A, Striano P, Iapadre G, Zagaroli L, Bonanni P, Coppola G, et al. The pharmacological management of Lennox-Gastaut syndrome and critical literature review. Seizure. (2018) 63:17–25. doi: 10.1016/j.seizure.2018.10.016 PubMed Abstract | CrossRef Full Text | Google Scholar
- Verrotti A, Striano P. Novel therapeutic options for Dravet and Lennox-Gastaut syndrome. Expert Rev Neurother. (2021) 11:1–4. doi: 10.1080/14737175.2020.1862651 PubMed Abstract | CrossRef Full Text | Google Scholar
- Brigo F, Jones K, Eltze C, Matricardi S. Anti-seizure medications for Lennox-Gastaut syndrome. Cochrane Database Syst Rev. (2021) 4:CD003277. doi: 10.1002/14651858.CD003277.pub4 CrossRef Full Text | Google Scholar
- Kwan P, Brodie MJ. Epilepsy after the first drug fails: substitution or add-on? Seizure. (2000) 9:464–8. doi: 10.1053/seiz.2000.0442 PubMed Abstract | CrossRef Full Text | Google Scholar
- Louis EKS. Truly “Rational” polytherapy: maximizing efficacy and minimizing drug interactions, drug load, and adverse effects. Curr Neuro Pharmacol. (2009) 7:96–105. doi: 10.2174/157015909788848929 PubMed Abstract | CrossRef Full Text | Google Scholar
- Brodie MJ. Medical therapy of epilepsy: when to initiate treatment and when to combine? J Neurol. (2005) 252:125–30. doi: 10.1007/s00415-005-0735-x PubMed Abstract | CrossRef Full Text | Google Scholar
- Shi JJ, Whitlock JB, Chimato N, Vargas E, Karceski SC, Frank RD. Epilepsy treatment in adults and adolescents: expert opinion, 2016. Epilepsy Behav. (2017) 69:186–222. doi: 10.1016/j.yebeh.2016.11.018 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lee BI, Park KM, Kim SE, Heo K. Clinical opinion: earlier employment of polytherapy in sequential pharmacotherapy of epilepsy. Epilepsy Res. (2019) 156:106–65. doi: 10.1016/j.eplepsyres.2019.106165 PubMed Abstract | CrossRef Full Text | Google Scholar
- López González FJ, Rodríguez Osorio X, Gil-Nagel Rein A, Carreño Martínez M, Serratosa Fernández J, Villanueva Haba V, et al. Drug-resistant epilepsy: definition and treatment alternatives. Neurologia. (2015) 30:439–46. doi: 10.1016/j.nrleng.2014.04.002 PubMed Abstract | CrossRef Full Text | Google Scholar
- French JA, French JA, Krauss GL, Biton V, Squillacote D, Yang H, et al. Adjunctive perampanel for refractory partial-onset seizures: randomized phase III study 304. Neurology. (2012) 79:589–96. doi: 10.1212/WNL.0b013e3182635735 PubMed Abstract | CrossRef Full Text | Google Scholar
- French JA, Krauss GL, Steinhoff BJ, Squillacote D, Yang H, Kumar D, et al. Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia. (2013) 54:117–25. doi: 10.1111/j.1528-1167.2012.03638.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Krauss GL, Serratosa JM, Villanueva V, Endziniene M, Hong Z, French J, et al. Randomized phase III study 306: adjunctive perampanel for refractory partial-onset seizures. Neurology. (2012) 78:1408–15. doi: 10.1212/WNL.0b013e318254473a PubMed Abstract | CrossRef Full Text | Google Scholar
- Krauss GL, Perucca E, Ben-Menachem E, Kwan P, Shih JJ, Clément JF, et al. Long-term safety of perampanel and seizure outcomes in refractory partial-onset seizures and secondarily generalized seizures: results from phase III extension study 307. Epilepsia. (2014) 55:1058–68. doi: 10.1111/epi.12643 PubMed Abstract | CrossRef Full Text | Google Scholar
- Krauss GL, Perucca E, Kwan P, Ben-Menachem E, Wang XF, Shih JJ, et al. Final safety, tolerability, and seizure outcomes in patients with focal epilepsy treated with adjunctive perampanel for up to 4 years in an open-label extension of phase III randomized trials: Study 307. Epilepsia. (2018) 59:866–76. doi: 10.1111/epi.14044 PubMed Abstract | CrossRef Full Text | Google Scholar
- French JA, Krauss GL, Wechsler RT, Wang XF, DiVentura B, Brandt C, et al. Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy a randomized trial. Neurology. (2015) 85:950–7. doi: 10.1212/WNL.0000000000001930 PubMed Abstract | CrossRef Full Text | Google Scholar
- Operto FF, Pastorino GMG, Mazza R, Di Bonaventura C, Matricardi S, Verrotti A, et al. Perampanel tolerability in children and adolescents with focal epilepsy: effects on behavior and executive functions. Epilepsy Behav. (2020) 103:106879. doi: 10.1016/j.yebeh.2019.106879 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lattanzi S, Cagnetti C, Foschi N, Provinciali L, Silvestrini M. Brivaracetam add-on for refractory focal epilepsy: a systematic review and meta-analysis. Neurology. (2016) 86:1344–52. doi: 10.1212/WNL.0000000000002545 PubMed Abstract | CrossRef Full Text | Google Scholar
- Verrotti A, Grasso EA, Cacciatore M, Matricardi S, Striano P. Potential role of brivaracetam in pediatric epilepsy. Acta Neurol Scand. (2021) 143:19–26. doi: 10.1111/ane.13347 PubMed Abstract | CrossRef Full Text | Google Scholar
- Contin M, Mohamed S, Santucci M, Lodi MAM, Russo E, Mecarelli O, et al. Cannabidiol in Pharmacoresistant Epilepsy: clinical pharmacokinetic data from an expanded access program. Front Pharmacol. (2021) 12:637801. doi: 10.3389/fphar.2021.637801 PubMed Abstract | CrossRef Full Text | Google Scholar
- Devinsky O, Cross JH, Laux L. Cannabidiol in Dravet syndrome study group. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. (2017) 376:2011–20. doi: 10.1056/NEJMoa1611618 CrossRef Full Text | Google Scholar
- Devinsky O, Patel AD, Thiele EA, Wong MH, Appleton R, Harden CL, et al. GWPCARE1 part a study group: randomized, dose ranging safety trial of cannabidiol in Dravet syndrome. Neurology. (2018) 90:1204–11. doi: 10.1212/WNL.0000000000005254 CrossRef Full Text | Google Scholar
- Devinsky O, Patel AD, Cross JH, Villanueva V, Wirrell EC, Privitera M, et al. GWPCARE3 study group: effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med. (2018) 378:1888–97. doi: 10.1056/NEJMoa1714631 CrossRef Full Text | Google Scholar
- Thiele EA, Marsh ED, French JA, Mazurkiewicz-Beldzinska M, Benbadis SR, Joshi C, et al. Cannabidiol in patients with seizures associated with Lennox-Gastautsyndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. (2018) 391:1085–96. doi: 10.1016/S0140-6736(18)30136-3 PubMed Abstract | CrossRef Full Text | Google Scholar
- Devinsky O, Nabbout R, Miller I, Laux L, Zolnowska M, Wright S, et al. Long-term cannabidiol treatment in patients with Dravet syndrome: an open-label extension trial. Epilepsia. (2019) 60:294–302. doi: 10.1111/epi.14628 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lattanzi S, Trinka E, Russo E, Striano P, Citraro R, Silvestrini M, et al. Cannabidiol as adjunctive treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome. Drugs Today. (2019) 55:177–96. doi: 10.1358/dot.2019.55.3.2909248 PubMed Abstract | CrossRef Full Text | Google Scholar
- Miller I, Scheffer IE, Gunning B, Sanchez-Carpintero R, Gil-Nagel A, Perry MS, et al. Dose-ranging effect of adjunctive oral cannabidiol vs placebo on convulsive seizure frequency in Dravet Syndrome: a randomized clinical trial. JAMA Neurol. (2020) 77:613–21. doi: 10.1001/jamaneurol.2020.0073 PubMed Abstract | CrossRef Full Text | Google Scholar
- Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. (2015) 56:1246–51. doi: 10.1111/epi.13060 PubMed Abstract | CrossRef Full Text | Google Scholar
- Devinsky O, Thiele EA, Wright S, Checketts D, Morrison G, Dunayevich E, et al. Cannabidiol efficacy independent of clobazam: meta-analysis of four randomized controlled trials. Acta Neurol Scand. (2020) 142:531–40. doi: 10.1111/ane.13305 PubMed Abstract | CrossRef Full Text | Google Scholar
- Anderson LL, Absalom NL, Abelev SV, Low IK, Doohan PT, Martin LJ, et al. Coadministered cannabidiol and clobazam: preclinical evidence for both pharmacodynamic and pharmacokinetic interactions. Epilepsia. (2019) 60:2224–34. doi: 10.1111/epi.16355 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lattanzi S, Trinka E, Striano P, Zaccara G, Del Giovane C, Nardone R, et al. Cannabidiol efficacy and clobazam status: a systematic review and meta-analysis. Epilepsia. (2020) 61:1090–8. doi: 10.1111/epi.16546 PubMed Abstract | CrossRef Full Text | Google Scholar
- Schoonjans AS, Lagae L, Ceulemans B. Low-dose fenfluramine in the treatment of neurologic disorders: experience in Dravet syndrome. Ther Adv Neurol Disord. (2015) 8:328–38. doi: 10.1177/1756285615607726 PubMed Abstract | CrossRef Full Text | Google Scholar
- Balagura G, Cacciatore M, Grasso EA, Striano P, Verrotti A. Fenfluramine for the treatment of Dravet syndrome and lennox-gastaut syndrome. CNS Drugs. (2020) 34:1001–7. doi: 10.1007/s40263-020-00755-z PubMed Abstract | CrossRef Full Text | Google Scholar
- Ceulemans B, Boel M, Leyssens K, Van Rossem C, Neels P, Jorens PG, et al. Successful use of fenfluramine as an add-on treatment for Dravet syndrome. Epilepsia. (2012) 53:1131–9. doi: 10.1111/j.1528-1167.2012.03495.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Schoonjans A, Paelinck BP, Marchau F, Gunning B, Gammaitoni A, Galer BS, et al. Low-dose fenfluramine significantly reduces seizure frequency in Dravet syndrome: a prospective study of a new cohort of patients. Eur J Neurol. (2017) 24:309–14. doi: 10.1111/ene.13195 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double-blind, placebo-controlled trial. Lancet. (2020) 394:2243–54. doi: 10.1016/S0140-6736(19)32500-0 PubMed Abstract | CrossRef Full Text | Google Scholar
- Nabbout R, Mistry A, Zuberi S, Villeneuve N, Gil-Nagel A, Sanchez-Carpintero R, et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: a randomized clinical trial. JAMA Neurol. (2019) 77:300–8. doi: 10.1001/jamaneurol.2019.4113 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lagae L, Schoonjans AS, Gammaitoni AR, Galer BS, Ceulemans B. A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox-Gastaut syndrome. Epilepsia. (2018) 59:1881–8. doi: 10.1111/epi.14540 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lai WW, Galer BS, Wong PC, Farfel G, Pringsheim M, Keane MG, et al. Cardiovascular safety of fenfluramine in the treatment of Dravet syndrome: analysis of an ongoing long-term open-label safety extension study. Epilepsia. (2020) 61:2386–95. doi: 10.1111/epi.16638 PubMed Abstract | CrossRef Full Text | Google Scholar
- Bialer M, Johannessen SI, Koepp MJ, Levy RH, Perucca E, Tomson T, et al. Progress report on new antiepileptic drugs: a summary of the Fourteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XIV). I. drugs in preclinical and early clinical development. Epilepsia. (2018) 59:1811–41. doi: 10.1111/epi.14557 PubMed Abstract | CrossRef Full Text | Google Scholar
- Krauss GL, Klein P, Brandt C, Lee SK, Milanov I, Milovanovic M, et al. Safety and efficacy of adjunctive cenobamate (YKP3089) in patients with uncontrolled focal seizures: a multicentre, double-blind, randomised, placebo-controlled, dose-response trial. Lancet Neurol. (2020) 19:38–48. doi: 10.1016/S1474-4422(19)30399-0 PubMed Abstract | CrossRef Full Text | Google Scholar
- Ragona F, Matricardi S, Castellotti B, Patrini M, Freri E, Binelli S, et al. Refractory absence epilepsy and glut1 deficiency syndrome: a new case report and literature review. Neuropediatrics. (2014) 45:328–32. doi: 10.1055/s-0034-1378130 PubMed Abstract | CrossRef Full Text | Google Scholar
- Perucca P, Perucca E. Identifying mutations in epilepsy genes: Impact on treatment selection. Epilepsy Res. (2019) 152:18–30. doi: 10.1016/j.eplepsyres.2019.03.001 PubMed Abstract | CrossRef Full Text | Google Scholar
- Wang J, Lin ZJ, Liu L, Xu HQ, Shi YW, Yi YH, et al. Epilepsy-associated genes. Seizure. (2017) 44:11–20. doi: 10.1016/j.seizure.2016.11.030 CrossRef Full Text | Google Scholar
- Simasathien T, Vadera S, Najm I, Gupta A, Bingama W, Jehi L. Improved outcomes with earlier surgery for intractable frontal lobe epilepsy. Ann Neurol. (2013) 73:646–54. doi: 10.1002/ana.23862 PubMed Abstract | CrossRef Full Text | Google Scholar
- Barba C, Cross JH, Braun K, Cossu M, Klotz KA, De Masi S, et al. Trends in pediatric epilepsy surgery in Europe between 2008 and 2015: country-, center-, and age-specific variation. Epilepsia. (2020) 61:216–27. doi: 10.1111/epi.16414 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lattanzi S, Cagnetti C, Matricardi S, Silvestrini M. Palliative non-resective surgery for drug-resistant epilepsy. Brain Dev. (2018) 40:512–3. doi: 10.1016/j.braindev.2017.12.012 PubMed Abstract | CrossRef Full Text | Google Scholar
- Boon P, De Cock E, Mertens A, Trinka E. Neurostimulation for drug-resistant epilepsy: a systematic review of clinical evidence for efficacy, safety, contraindications and predictors for response. Curr Opin Neurol. (2018) 31:198–210. doi: 10.1097/WCO.0000000000000534 PubMed Abstract | CrossRef Full Text | Google Scholar
- Starnes K, Miller K, Wong-Kisiel L, Lundstrom BN. A review of neurostimulation for epilepsy in pediatrics. Brain Sci. (2019) 9:283. doi: 10.3390/brainsci9100283 CrossRef Full Text | Google Scholar
- Pérez-Carbonell L, Faulkner H, Higgins S, Koutroumanidis M, Leschziner G. Vagus nerve stimulation for drug-resistant epilepsy. Pract Neurol. (2020) 20:189–98. doi: 10.1136/practneurol-2019-002210 CrossRef Full Text | Google Scholar
- Morris GL, Gloss D, Buchhalter J, Mack KJ, Nickels K, Harden C. Evidence-based guideline update: vagus nerve stimulation for the treatment of epilepsy: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. (2013) 81:1453–9. doi: 10.1212/WNL.0b013e3182a393d1 PubMed Abstract | CrossRef Full Text | Google Scholar
- Ben-Menachem E, Hamberger A, Hedner T, Hammond EJ, Uthman BM, Slater J, et al. Effects of vagus nerve stimulation on amino acids and other metabolites in the CSF of patients with partial seizures. Epilepsy Res. (1995) 20:221–7. doi: 10.1016/0920-1211(94)00083-9 PubMed Abstract | CrossRef Full Text | Google Scholar
- Fisher B, DesMarteau JA, Koontz EH, Wilks SJ, Melamed SE. Responsive vagus nerve stimulation for drug resistant epilepsy: a review of new features and practical guidance for advanced practice providers. Front Neurol. (2021) 11:610379. doi: 10.3389/fneur.2020.610379 CrossRef Full Text | Google Scholar
- Chambers A, Bowen JM. Electrical stimulation for drug-resistant epilepsy: an evidence-based analysis. Ont Health Technol Assess Ser. (2013) 13:1–37. PubMed Abstract | Google Scholar
- Panebianco M, Rigby A, Weston J, Marson AG. Vagus nerve stimulation for partial seizures. Cochrane Database Syst Rev. (2015) 2015:CD002896. doi: 10.1002/14651858.CD002896.pub2 CrossRef Full Text | Google Scholar
- Klinkenberg S, Aalbers MW, Vles JS, Cornips EM, Rijkers K, Leenen L, et al. Vagus nerve stimulation in children with intractable epilepsy: a randomized controlled trial. Dev Med Child Neurol. (2012) 54:855–61. doi: 10.1111/j.1469-8749.2012.04305.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Fisher R, Salanova V, Witt T, Worth R, Henry T, Gross R, et al. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. (2010) 51:899–908. doi: 10.1111/j.1528-1167.2010.02536.x PubMed Abstract | CrossRef Full Text | Google Scholar
- Salanova V, Witt T, Worth R, Henry TR, Gross RE, Nazzaro JM, et al. Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology. (2015) 84:1017–25. doi: 10.1212/WNL.0000000000001334 PubMed Abstract | CrossRef Full Text | Google Scholar
- Li MCH, Cook MJ. Deep brain stimulation for drug-resistant epilepsy. Epilepsia. (2018) 59:273–90. doi: 10.1111/epi.13964 CrossRef Full Text | Google Scholar
- Lim S-N, Lee C-Y, Lee S-T, Tu PH, Chang BL, Lee CH, et al. Low and high frequency hippocampal stimulation for drug-resistant mesial temporal lobe epi-lepsy. Neuromodulation. (2016) 19:365–72. doi: 10.1111/ner.12435 CrossRef Full Text | Google Scholar
- Valentin A, Garcia Navarrete E, Chelvarajah R, Torres C, Navas M, Vico L, et al. Deep brain stimulation of the centromedian thalamic nucleus for the treatment of generalized and frontal epilepsies. Epilepsia. (2013) 54:1823–33. doi: 10.1111/epi.12352 PubMed Abstract | CrossRef Full Text | Google Scholar
- Matias CM, Sharan A, Wu C. Responsive neurostimulation for the treatment of epilepsy. Neurosurg Clin N Am. (2019) 30:231–42. doi: 10.1016/j.nec.2018.12.006 CrossRef Full Text | Google Scholar
- Bergey GK, Morrell MJ, Mizrahi EM, Goldman A, King-Stephens D, Nair D, et al. Long-term treatment with responsive brain stimulation in adults with refractory partial seizures. Neurology. (2015) 84:810–7. doi: 10.1212/WNL.0000000000001280 PubMed Abstract | CrossRef Full Text | Google Scholar
- Geller EB, Skarpaas TL, Gross RE, Goodman RR, Barkley GL, Bazil CW, et al. Brain responsive neurostimulation in patients with medically intractable mesial temporal lobe epilepsy. Epilepsia. (2017) 58:994–1004. doi: 10.1111/epi.13740 PubMed Abstract | CrossRef Full Text | Google Scholar
- Nair DR, Laxer KD, Weber PB, Murro AM, Park YD, Barkley GL, et al. Nine-year prospective efficacy and safety of brain-responsive neurostimulation for focal epilepsy. Neurology. (2020) 95:e1244–56. doi: 10.1212/WNL.0000000000010154 PubMed Abstract | CrossRef Full Text | Google Scholar
- Heck CN, King-Stephens D, Massey AD, Nair DR, Jobst BC, Barkley GL, et al. Two year seizure reduction in adults with medically intractable partial onset epilepsy treated with responsive neurostimulation: final results of the RNS system pivotal trial. Epilepsia. (2014) 55:432–41. doi: 10.1111/epi.12534 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lundstrom BN, Worrell GA, Stead M, Van Gompel JJ. Chronic subthreshold cortical stimulation: a therapeutic and potentially restorative therapy for focal epilepsy. Expert Rev Neurother. (2017) 17:661–6. doi: 10.1080/14737175.2017.1331129 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lefaucheur JP, André-Obadia N, Antal A, Ayache SS, Baeken C, Benninger DH, et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS). Clin Neurophysiol. (2014) 125:2150–206. doi: 10.1016/j.clinph.2014.05.021 PubMed Abstract | CrossRef Full Text | Google Scholar
- Lefaucheur JP, Aleman A, Baeken C, Benninger DH, Brunelin J, Di Lazzaro V, et al. Evidence-based guidelines on the therapeutic use of repetitive transcranial magnetic stimulation (rTMS): an update (2014-2018). Clin Neurophysiol. (2020) 131:474–528. doi: 10.1016/j.clinph.2020.02.003 PubMed Abstract | CrossRef Full Text | Google Scholar
- Fregni F, El-Hagrassy MM, Pacheco-Barrios K, Carvalho S, Leite J, Simis M, et al. Evidence-based guidelines and secondary meta-analysis for the use of transcranial direct current stimulation (tDCS) in neurological and psychiatric disorders. Int J Neuropsychopharmacol. (2020) 26:pyaa051. doi: 10.1093/ijnp/pyaa051 CrossRef Full Text | Google Scholar
- Yang D, Wang Q, Xu C, Fang F, Fan J, Li L, et al. Transcranial direct current stimulation reduces seizure frequency in patients with refractory focal epilepsy: a randomized, double-blind, sham-controlled, and three-arm parallel multicenter study. Brain Stimul. (2020) 13:109–16. doi: 10.1016/j.brs.2019.09.006 PubMed Abstract | CrossRef Full Text | Google Scholar
- Verrotti A, Iapadre G, Di Francesco L, Zagaroli L, Farello G. Diet in the treatment of epilepsy: what we know so far. Nutrients. (2020) 12:2645. doi: 10.3390/nu12092645 PubMed Abstract | CrossRef Full Text | Google Scholar
- Operto FF, Matricardi S, Pastorino GMG, Verrotti A, Coppola G. The ketogenic diet for the treatment of mood disorders in comorbidity with epilepsy in children and adolescents. Front Pharmacol. (2020) 11:578396. doi: 10.3389/fphar.2020.578396 PubMed Abstract | CrossRef Full Text | Google Scholar
- Martin-McGill KJ, Bresnahan R, Levy RG, Cooper PN. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev. (2020) 6:CD001903. doi: 10.1002/14651858.CD001903.pub5 CrossRef Full Text | Google Scholar
Informations sur le droit d’auteur
Article publié sous les conditions définies par la licence Creative Commons Attribution License CC-BY, qui autorise sans restrictions l’utilisation, la diffusion, et la reproduction sur quelque support que ce soit, sous réserve de citation correcte de la publication originale.
Voir la publication originale
Fattorusso A, Matricardi S, Mencaroni E, Dell’Isola GB, Di Cara G, Striano P and Verrotti A (2021). The Pharmacoresistant Epilepsy: An Overview on Existant and New Emerging Therapies. Front. Neurol. 12:674483. doi: 10.3389/fneur.2021.674483

{kind=link}