 sont des loci spéciaux de bactéries et d'archées, constitués de séquences répétitives directes séparées par des séquences uniques (espaceurs). Les espaceurs sont empruntés à des éléments génétiques étrangers rencontrés par la cellule (bactériophages, plasmides). L'ARN transcrit à partir des loci CRISPR, ainsi que les protéines Cas associées, fournissent une immunité adaptative grâce à la liaison complémentaire de l'ARN aux acides nucléiques d'éléments étrangers et à leur destruction ultérieure par les protéines Cas. Cependant, à ce jour, il existe de nombreuses preuves de l'implication de CRISPR dans des processus non liés à l'immunité. L’utilisation des techniques CRISPR-Cas[de] pour l’édition dirigée du génome est une direction prometteuse du génie génétique moderne. Pour 2016, les scientifiques utilisent largement des approches basées sur les systèmes CRISPR-Cas ; peut-être qu’à l’avenir, ces approches seront utilisées en médecine pour le traitement des maladies héréditaires. En outre, CRISPR-Cas est important pour l'administration ciblée de médicaments et leur libération sous influence externe - pour cela, des matériaux contenant des coupes d'ADN sont utilisés.){kind=link}
Les CRISPR (clustered Regular Interspaced Short Palindromic Repeats – courtes répétitions palindromiques régulièrement disposées en groupes) sont des loci spéciaux de bactéries et d’archées, constitués de séquences répétitives directes séparées par des séquences uniques (espaceurs). Les espaceurs sont empruntés à des éléments génétiques étrangers rencontrés par la cellule (bactériophages, plasmides). L’ARN transcrit à partir des loci CRISPR, ainsi que les protéines Cas associées, fournissent une immunité adaptative grâce à la liaison complémentaire de l’ARN aux acides nucléiques d’éléments étrangers et à leur destruction ultérieure par les protéines Cas. A ce jour, il existe de nombreuses preuves de l’implication de CRISPR dans des processus non liés à l’immunité. L’utilisation des techniques CRISPR-Cas pour l’édition dirigée du génome est une direction prometteuse du génie génétique moderne. Depuis 2016, les scientifiques utilisent largement des approches basées sur les systèmes CRISPR-Cas ; peut-être qu’à l’avenir, ces approches seront utilisées en médecine pour le traitement des maladies héréditaires. En outre, CRISPR-Cas est important pour l’administration ciblée de médicaments et leur libération sous influence externe – pour cela, des matériaux contenant des coupes d’ADN sont utilisés.
Histoire des études
Le premier locus CRISPR a été découvert dans la bactérie Escherichia coli en 1987 par un groupe de scientifiques japonais dirigé par Yoshizumi Isino. Ils ont remarqué des éléments répétitifs dans le génome de cette bactérie, séparés par des séquences non répétitives (espaceurs) ; cependant, les scientifiques n’attachaient pas beaucoup d’importance à leur observation. Une étude à grande échelle de CRISPR a été lancée par le chercheur espagnol Francisco Mojica, qui a découvert en 1993 des séquences répétées séparées par des lacunes dans le génome de l’archéen Haloferax mediterranei. Il a remarqué que les répétitions dans les génomes de cette archée et de E. coli ont une structure très similaire, mais n’ont rien de commun dans les séquences nucléotidiques. Selon Mojic, des répétitions de structure si similaire, qui sont systématiquement des groupes de procaryotes très éloignés, doivent remplir une fonction très importante. Initialement, il a appelé la nouvelle classe de répétitions «courtes répétitions régulièrement espacées, SRSR», mais plus tard, à sa suggestion, ce nom a été changé en «courtes répétitions palindromiques régulièrement disposées en groupes» (clustered Regular Interspaced Short Palindromic Repeats, CRISPR). Mojica a continué à rechercher CRISPR dans le génome d’autres microbes et, en 2000, il les avait trouvés dans 20 micro-organismes, dont le bacille de la peste Yersinia pestis et d’autres agents pathogènes. En 2002, les gènes cas, les gènes des locus CRISPR codant pour les protéines Cas, ont été découverts.
Malgré la découverte des systèmes CRISPR-Cas chez une grande variété de procaryotes, on savait peu de choses sur les fonctions de CRISPR jusqu’en 2005. En 2005, Mojica et ses collègues ont publié les résultats de leurs nouvelles recherches, dans lesquels ils ont découvert que les espaceurs correspondaient à des séquences du génome des bactériophages, ainsi qu’à des sections de plasmides. Ils ont également constaté que les souches d‘E. coli dont les locus CRISPR contiennent un espaceur correspondant au phage P1 sont résistantes à ce phage et ont conclu que les locus CRISPR sont associés à l’immunité adaptative des procaryotes. La même année, deux autres groupes (Pourcel C. et al., et Bolotin et al.) de recherche ont publié des publications parvenues à la même conclusion.
En 2006, une classification des CRISPR connus a été développée et un mécanisme possible d’immunité adaptative basée sur CRISPR a été proposé. En 2007, un groupe de recherche dirigé par Philip Horvath a finalement établi et prouvé expérimentalement la participation de CRISPR pour assurer le travail de l’immunité adaptative spécifique aux séquences cibles; en même temps, le rôle clé des protéines Cas dans ce processus a été révélé. Pour cette réalisation, il a reçu le prix Massry en 2015, aux côtés d’autres scientifiques ayant apporté une contribution significative à l’étude de CRISPR (Jennifer Dowdna et Emmanuelle Charpentier). En 2008, il a été démontré que le système CRISPR nécessite un ARN CRISPR spécialement traité (ARNcr) pour fonctionner, et la capacité du système CRISPR à effectuer des interférences avec l’ADN a également été démontrée. L’interférence, le ciblage des ARN et le ciblage contre des séquences d’ADN spécifiques sont trois découvertes réalisées en 2007-2008 qui ont lancé le développement de méthodes de génie génétique basées sur CRISPR.
Un certain nombre de découvertes importantes ultérieures concernant la conception de systèmes CRISPR de type II (notamment la nécessité de la protéine Cas9 et d’un petit ARN supplémentaire, appelé tracrRNA, en plus du crRNA), ont permis en 2012 de tester expérimentalement le premier système CRISPR de type II développé artificiellement. Début 2013 (à environ deux semaines d’intervalle), plusieurs groupes ont montré que les systèmes artificiels CRISPR-Cas pouvaient fonctionner non seulement dans les cellules bactériennes et in vitro, mais également dans les cellules eucaryotes. En 2012, le biochimiste lituanien Virginijus Šikšnis a été l’un des premiers à démontrer le clivage programmé de l’ADN par l’un des composants des systèmes CRISPR-Cas par la protéine Cas9. Depuis 2007, ses recherches portent principalement sur l’étude des systèmes CRISPR-Cas récemment découverts pour protéger les bactéries contre les bactériophages et le matériel génétique étranger. Selon Šikšnis, son article n’a même pas été reconnu par un comité de rédaction sérieux d’une revue académique et n’a pas été envoyé aux évaluateurs, de sorte que le temps nécessaire pour le reconnaître comme le premier a été perdu. Martin Szlak a rapporté que Šikšnis avait soumis son article décrivant le clivage de l’ADN avec Cas9 dans la revue scientifique à comité de lecture Cell Reports le 18 avril 2012. Après qu’il ait été rejeté sans examen par les pairs, il l’a soumis un mois plus tard à la revue scientifique à comité de lecture PNAS et a mis plusieurs mois à l’examiner et à le publier. Entre-temps, la biochimiste américaine Jennifer Downa et la microbiologiste française Emmanuelle Charpentier ont publié leur article dans la revue scientifique à comité de lecture Science, où il a été examiné et accepté en deux semaines. Par la suite, la technologie d’édition du génome Cas9 a obtenu une licence de DuPont.
Les deux années et demie suivantes ont vu le développement des méthodes CRISPR et l’application de cette méthode à divers groupes d’organismes. En avril 2015, un groupe de scientifiques chinois a publié les résultats de leur étude dans laquelle les génomes d’embryons humains ont été modifiés à l’aide de CRISPR-Cas9. Cependant, la précision du montage dans cette expérience était très faible et l’expérience elle-même a été perçue de manière ambiguë par la communauté scientifique. Début 2016, des scientifiques américains ont annoncé avoir réussi à réduire à presque zéro le nombre d’erreurs dans le fonctionnement de CRISPR-Cas9. La même année, un groupe de scientifiques a découvert que les systèmes CRISPR-Cas provenaient de transposons ayant perdu leur mobilité et fixés dans le génome. Une étude phylogénétique a montré que toutes les endonucléases Cas descendent d’un seul transposon de tous les transposons portant l’endonucléase IscB.
En 2012 et 2013, au début de la percée de CRISPR dans le génie génétique, la méthode CRISPR-Cas a été nominée pour le prix Breakthrough of the Year décerné par Science Magazine. En 2015, il a remporté ce prix. À ce jour, CRISPR est considérée comme l’innovation technologique la plus importante dans les sciences de la vie depuis l’invention de la réaction en chaîne par polymérase (PCR), découverte trois décennies plus tôt. Pour l’introduction des techniques d’édition génétique à l’aide de CRISPR-Cas9, Jennifer Doudna et Emmanuelle Charpentier ont reçu le prix Nobel de chimie 2020.
Principes généraux
Les systèmes CRISPR-Cas diffèrent à la fois structurellement et fonctionnellement. Cependant, tous les systèmes CRISPR-Cas présentent un certain nombre de caractéristiques communes.
Les locus CRISPR ne peuvent remplir la fonction d’immunité qu’en présence de gènes cas, qui sont généralement situés à proximité immédiate de CRISPR. L’ensemble des gènes cas détermine le type de système CRISPR-Cas. Les loci CRISPR sont de courtes répétitions directes (généralement environ 30 à 40 nucléotides de long) qui sont séparées les unes des autres par des espaceurs non répétitifs dérivés de l’ADN des éléments génétiques étrangers rencontrés par la cellule ou ses précurseurs. La longueur des espaceurs est généralement comparable à la longueur des répétitions. Avant un certain nombre de répétitions et d’espaceurs, il existe une séquence leader contenant, en règle générale, un promoteur à partir duquel commence la transcription unidirectionnelle des répétitions et des espaceurs CRISPR. Les espaceurs sont entièrement intégrés au génome cellulaire et sont transmis à ses descendants lors de la division cellulaire. Chez les bactéries, l’intégration de nouveaux espaceurs dans le génome se combine avec la perte de gènes redondants et étrangers ; les bactéries parviennent ainsi à éviter une augmentation significative de la taille du génome, contrairement aux eucaryotes supérieurs, chez lesquels des séquences répétitives dérivées d’éléments génétiques exogènes constituent une partie essentielle du génome.
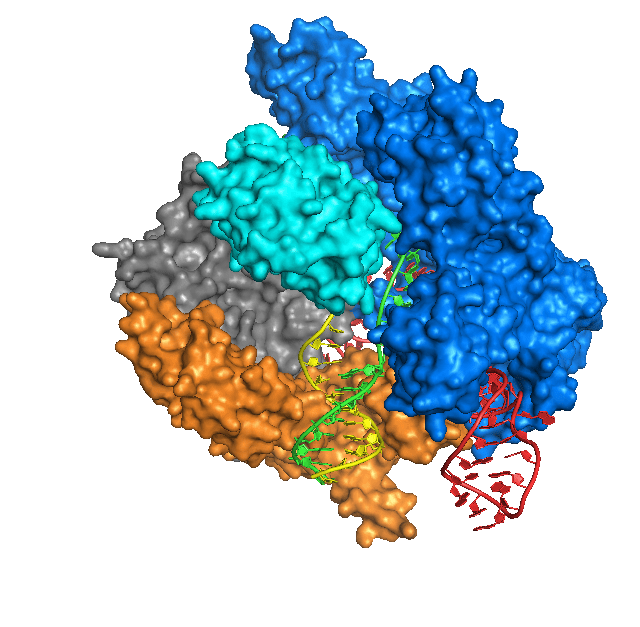
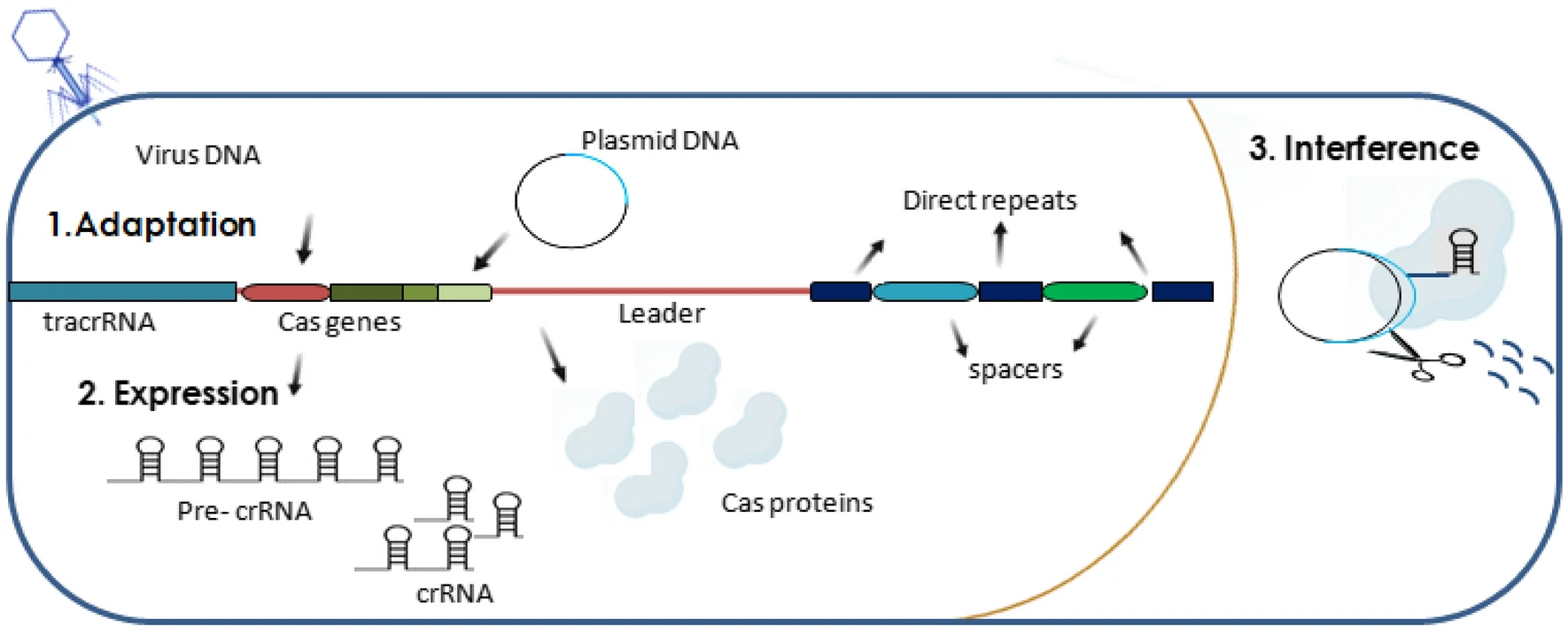
En plus de la similarité structurelle, divers systèmes CRISPR-Cas combinent trois étapes clés de l’immunité médiée par CRISPR : l’acquisition ou l’adaptation, l’expression et l’interférence. Au stade de l’acquisition, un nouvel espaceur est inséré dans CRISPR, formé à partir d’un élément génétique étranger entré dans la cellule. Au stade de l’expression, la transcription CRISPR et le traitement des ARN CRISPR courts (ARNcr) ciblant une cible spécifique ont lieu. Lors de l’interférence, le complexe ribonucléoprotéique crRNA-Cas reconnaît l’acide nucléique cible en raison de l’appariement de bases complémentaires de la cible avec le crRNA, après quoi il coupe la cible en raison de l’activité endonucléase et/ou exonucléase des protéines Cas. Il est intéressant de noter que le travail des systèmes CRISPR-Cas présente de nombreux points fondamentaux en commun avec le travail du système immunitaire des mammifères. Ainsi, même un bactériophage défectueux peut provoquer une immunisation CRISPR (c’est-à-dire l’insertion d’un nouvel espaceur), tout comme une réponse immunitaire chez un mammifère peut se développer lorsqu’un agent pathogène tué est introduit.
Les systèmes CRISPR-Cas peuvent être transférés d’un micro-organisme à l’autre par transfert horizontal de gènes. Lutter contre l’invasion d’éléments génétiques étrangers dans une bactérie n’est pas toujours bénéfique pour la bactérie. Par exemple, la bactérie Staphylococcus epidermidis peut connaître une diminution de la résistance aux antibiotiques en raison de la destruction des plasmides conjugatifs qui ont fourni cette résistance par le système CRISPR-Cas. Chez Staphylococcus aureus, un nombre réduit de loci CRISPR entraîne une augmentation du nombre de prophages, de plasmides et d’éléments génétiques mobiles dans la cellule, ce qui renforce la virulence de la bactérie. Cependant, les loci CRISPR-Cas, qui empêchent la propagation d’éléments génétiques mobiles utiles dans des conditions données, pourraient disparaître.
Achat d’entretoises
Étant donné que l’immunité adaptative médiée par CRISPR est codée dans l’ADN, le processus d’immunisation implique la copie et l’insertion d’éléments génétiques étrangers dans CRISPR en tant que nouveaux espaceurs. Les espaceurs constituent la mémoire immunologique, qui stocke des informations sur les infections passées, et c’est elle qui est à la base de la réponse à l’invasion répétée d’éléments génétiques similaires. La plupart des données sur les mécanismes moléculaires d’acquisition de nouveaux espaceurs ont été obtenues en étudiant le système CRISPR d’Escherichia coli de type I et de Streptococcus thermophilus de type II. L’orientation correcte et l’insertion d’un nouvel espaceur se produisent avec la participation de la séquence immédiatement au-dessus de la première répétition ; ainsi, de nouveaux espaceurs sont ajoutés à l’extrémité 5′ du locus CRISPR. L’intégration du nouvel espaceur entre la séquence leader et la première répétition est réalisée par le complexe Cas1-Cas2-protospacer. Dans certains systèmes CRISPR-Cas, des protéines supplémentaires sont impliquées dans ce processus. Lorsqu’un nouvel espaceur est inséré, une duplication de la répétition se produit, grâce à laquelle la structure correcte du locus est préservée, qui doit commencer par une répétition.
Étant donné que les espaceurs sont transmis des ancêtres aux descendants au cours de la division cellulaire, en présence d’espaceurs similaires, des relations phylogénétiques peuvent être établies entre des souches ayant des espaceurs ancestraux communs, ainsi que des souches possédant de nouveaux espaceurs récemment acquis.
Dans les systèmes de type I et II, l’insertion d’un espaceur ne peut se produire qu’à partir des éléments étrangers dans lesquels une séquence spéciale PAM (protospacer adjacent motif) est adjacente au protospacer. De plus, la bactérie doit distinguer le matériel génétique étranger du sien afin de ne pas insérer un fragment de son propre chromosome comme espaceur et de ne pas cibler le système CRISPR-Cas sur son génome, ce qui serait fatal pour la cellule. Le système CRISPR-Cas d’E. coli type I distingue son ADN par la présence de Chi-sites — des motifs à 8 nucléotides qui se répètent dans son génome en moyenne toutes les 5 000 paires de bases. Bien que de nombreux espaceurs puissent être formés à partir du même élément génétique étranger, dans un élément génétique, certains motifs sont plus préférables lors du choix d’un futur espaceur. Probablement, ces motifs ont été déterminés à la suite d’une sélection naturelle associée à l’efficacité des espaceurs. Ainsi, certains espaceurs donnent naissance à des ARNcr qui ciblent les protéines Cas vers des séquences partiellement complémentaires.
Lorsqu’elles sont confrontées au même phage, différentes cellules inséreront des fragments légèrement différents de son génome comme espaceur, de sorte que de grandes populations possédant une grande variété d’espaceurs contre le même phage offrent une résistance plus efficace : si le phage mute de telle sorte que l’un des phages existants les espaceurs dans une population deviennent inefficaces, d’autres continueront à assurer la protection.
Expression et formation d’ARNcr
Après intégration dans CRISPR de parties d’éléments génétiques étrangers, il est nécessaire de les transformer sous une forme capable de cibler les protéines Cas pour cibler des séquences en vue de leur reconnaissance et de leur destruction. Cette forme est le crRNA guide, qui contient une séquence unique complémentaire d’une cible spécifique. Tout d’abord, un certain nombre de répétitions et d’espaceurs CRISPR sont transcrits en un seul long transcrit, le pré-ARNc, qui est ensuite découpé en courts ARNc. La plupart des répétitions dans CRISPR sont des palindromes, de sorte que leurs régions pré-ARNc correspondantes forment des épingles à cheveux. Dans de nombreux cas, ce sont ces épingles à cheveux qui sont reconnues par les protéines Cas qui transforment le pré-ARNc en ARNcr.
En règle générale, la transcription CRISPR dépend de la séquence leader et se produit en continu mais à un rythme lent. Cependant, ce taux augmente considérablement dans des conditions de stress ou lorsque la cellule entre en collision avec des phages, lui offrant ainsi une protection rapide et efficace. Des éléments promoteurs ont été trouvés non seulement dans la séquence leader, mais également dans les répétitions. Bien qu’un locus entier puisse être transcrit en une seule fois, il a été démontré que certains espaceurs d’un locus sont transcrits plus fréquemment que d’autres, comme les premiers espaceurs suivant la séquence leader et la première répétition. En effet, il est bien plus bénéfique pour une cellule de disposer d’une défense plus forte contre les éléments invasifs qu’elle a rencontrés dans un passé récent que contre ceux qu’elle a rencontrés il y a longtemps.
Ingérence
Au stade de l’interférence, les ARNc se lient à leurs cibles par appariement de bases et dirigent ainsi les endonucléases Cas pour couper et détruire la cible. La formation d’un complexe de protéines crRNA et Cas assure la destruction endonucléolytique des séquences complémentaires de crRNA NA. Bien que les cibles soient principalement de l’ADN double brin (ADNdb), certains systèmes CRISPR-Cas peuvent dégrader les ARN simple brin complémentaires (ARNsb). Les systèmes CRISPR-Cas qui reconnaissent l’ADNdb sont exigeants vis-à-vis des séquences adjacentes au protospacer : en particulier, dans les systèmes de type I et II, seules les cibles contenant le motif PAM sont reconnues (l’exigence de présence de PAM peut servir à se protéger contre la coupure le génome cellulaire par le système CRISPR-Cas). Les systèmes qui fonctionnent avec le ssRNA n’ont pas de telles exigences. Après l’attaque endonucléolytique initiale (casse la cible) par Cas, une destruction supplémentaire de la cible peut se produire sous l’action d’autres nucléases.
Variété de systèmes CRISPR-Cas
Tous les systèmes CRISPR-Cas connus peuvent être divisés en deux classes principales, 5 types et 16 sous-types, basés sur la présence ou l’absence de certains gènes cas, la structure de l’opéron cas, les séquences d’acides aminés des protéines Cas et les mécanismes qui assurer le fonctionnement de l’immunité médiée par CRISPR. À cela s’ajoute le système CRISPR-Rx (CRISPR-CasRx), qui cible l’ARN (contrairement aux autres CRISPR, notamment CRISPR-Cas9, qui cible l’ADN). Pour cette raison, CRISPR-Rx peut supprimer l’expression des gènes alors que le code génétique reste inchangé.
Les systèmes de première classe sont caractérisés par des complexes effecteurs multiprotéiques (Cascade, Cmr, Csm). Cette classe comprend les systèmes des types I, III et IV. Les systèmes de type I sont les systèmes CRISPR-Cas les plus courants. Leurs cibles sont l’ADNdb contenant le motif PAM, et la destruction est réalisée par le complexe multiprotéique effecteur Cascade associé à la protéine Cas3. Les systèmes de type III sont courants chez les archées et sont caractérisés par des complexes multiprotéiques Csm et Cmr. Ils peuvent reconnaître à la fois l’ADN et l’ARN, et la reconnaissance de l’ADN ne nécessite pas de PAM. Dans les systèmes de ce type, la destruction des cibles est réalisée par la protéine Cas10 en collaboration avec des nucléases effectrices, à savoir Cmr4 dans le sous-type IIIA (RNase, qui fait partie du complexe Cmr) et Csm3 dans le sous-type IIIB (RNase, qui fait partie du complexe Cmr). Les systèmes de type IV sont assez rares et leur distribution et leur mécanisme d’action ne sont pas bien compris.
Les systèmes de classe II ont une seule protéine effectrice. Cette classe comprend les types II et V. Les systèmes de type II sont activement utilisés en génie génétique ; ils sont caractérisés par la présence de l’endonucléase Cas9. Dans les systèmes de ce type, l’ARN guide n’est pas un ARNcr, mais un duplex d’ARNcr et un ARN supplémentaire, tracrARN. Le duplex crRNA:tracrRNA ordonne aux domaines nicase RuvC et HNH Cas9 d’introduire des cassures aux extrémités franches dans l’ADN cible, qui devraient avoir un PAM près de l’extrémité 3′. Les systèmes de type V sont rares et se caractérisent par la présence de la nucléase Cpf1, qui est dirigée par l’ARNc vers l’ADN cible. Cette nucléase de type RuvCeffectue une coupure sur un site distal par rapport à l’extrémité 3′ du PAM. Contrairement à Cas9, cette nucléase coupe l’ADNdb avec formation d’extrémités collantes, plutôt que émoussées, longues de 5 nucléotides.
Systèmes de types I et III
Comme mentionné ci-dessus, les systèmes de type I et de type III utilisent des complexes effecteurs multiprotéiques. Ils sont également unis par l’utilisation de la protéine Cas6 pour le traitement du pré-ARNc (parfois elle est remplacée par un orthologue, Cas5). Ces similitudes et quelques autres entre les systèmes de type I et III plaident en faveur de leur descendance d’un ancêtre commun. Les systèmes de type I sont subdivisés en six sous-types (I-A, I-B, I-C, I-D, I-E, I-F) sur la base des séquences d’acides aminés des protéines du complexe effecteur et de l’arrangement mutuel de leurs gènes (synthénie). Le système des sous-types I-E d’E. coli est le plus étudié.
Dans les systèmes de type I, le complexe effecteur – Cascade – inclut Cas6 en tant que sous-unité intégrale, de sorte que le traitement de l’ARNcr se produit au sein du complexe effecteur et que l’ARNcr mature y reste associé. Le complexe recherche alors sa séquence cible ; dans le même temps, il reconnaît probablement d’abord son PAM et seulement après cela, il vérifie les positions clés du protospacer pour la complémentarité des ARNcr. Puisqu’il n’y a pas de PAM dans les répétitions CRISPR, le génome d’une bactérie dotée d’un système CRISPR-Cas de type I est protégé de manière fiable contre la destruction par ce système. Lors de la liaison à Cascade, le protospacer forme une boucle R dans l’ADNdb cible, ce qui nécessite un super-enroulement négatif ; cela facilite probablement le déroulement de l’ADN indépendamment des nucléotides triphosphates (NTP). Le complexe Cascade-protospacer est reconnu par la protéine Cas3. Cas3 possède un domaine nucléase HD, ainsi qu’un domaine de déroulement-translocation qui nécessite des NTP pour son fonctionnement. Cas3 peut dérouler les duplex ADN:ADN et ADN:ARN. Le domaine HD est généralement situé à l’extrémité N de Cas3. Le domaine HD introduit une entaille dans la cible près du PAM, après quoi Cas3 se détache de Cascade et utilise son domaine d’hydrolyse de nucléoside triphosphate pour se déplacer plus loin le long de l’ADN, introduisant des entailles supplémentaires en cours de route.
La structure en cascade (libre et liée à l’ADN) d’E. coli a été visualisée à une résolution proche de l’atome. La cible est reconnue par l’appariement de bases Watson-Crick, bien qu’un nucléotide protospaceur sur six ne soit pas complémentaire du nucléotide crRNA correspondant. À cet égard, la géométrie globale du complexe ADN-crRNA ne correspond pas à une double hélice : les tours répétitifs en demi-hélice du duplex sont interrompus par des bases non appariées, ce qui permet à l’ADN de se replier sur le crRNA sans s’enrouler autour de lui. La liaison en cascade à la cible et à sa séquence associée présente des caractéristiques cinétiques et structurelles différentes, ce qui permet au complexe de faire la distinction entre la cible et les séquences proches de celle-ci. Dans le premier cas, il s’ensuit une interférence et une destruction de la cible, et dans le second, l’insertion d’un nouvel espaceur. Une telle adaptation dirigée, contrairement à l’adaptation primaire «naïve», nécessite le travail non seulement des protéines Cas1 et Cas2, mais également de Cas3.
En plus des 6 sous-types de systèmes de type I (I-A – I-F), un autre sous-type est connu, I-U (U de l’anglais uncharacterized), puisque le mécanisme de coupure du pré-ARNc et l’architecture du complexe effecteur lui sont inconnus. Contrairement à la plupart des systèmes de type I, la protéine I – U Cas3 possède un domaine HD à l’extrémité C-terminale.
Type III
Les systèmes de type III sont divisés en deux sous-types : III-A et III-B. Ils se caractérisent par la présence de la protéine Cas10, la plus grande sous-unité du complexe effecteur Csm (dans le cas du sous-type III-A) et Cmr (dans le cas du sous-type III-B). De plus, tous les systèmes de type III codent pour une protéine Cas5 et, en règle générale, pour plusieurs protéines Cas7 paralogues. Les deux sous-types sont caractérisés par l’utilisation de l’orthologue Cas6 pour le traitement du pré-ARNcr, bien que l’enzyme de traitement ne soit pas toujours un composant stable du complexe effecteur correspondant (comme dans les systèmes de type I). En 2008, il a été démontré que le système III-A de Staphylococcus epidermidis fonctionnait avec des cibles ADN, et en 2009, le système III-B de Pyrococcus furiosus fonctionnait avec l’ARN. Les systèmes III-A et III-B ne nécessitent pas la présence du motif PAM pour une reconnaissance réussie de la cible.
Une étude plus approfondie des systèmes de type III a révélé de nouveaux mystères dans la spécificité du substrat des sous-types III-A et III-B. Ainsi, il s’est avéré que le système III-A de S. epidermidis ne peut fonctionner qu’avec des protospaceurs transcrits. De plus, il s’est avéré que les complexes Csm de S. thermophilus et Thermus thermophilus ont une activité latente de dégradation de l’ARN et introduisent des cassures dans l’ARN tous les 6 nucléotides. La même activité a également été démontrée pour les complexes Cmr. Le système III-A de S. epidermidis détruit non seulement les transcrits synthétisés, mais coupe également l’ADN cible de manière dépendante de la transcription, au détriment des résidus d’acides aminés Cas10 spécifiques qui ne sont pas associés à la reconnaissance de la cible. L’hydrolyse de l’ARN médiée par les complexes Csm et Cmr n’est pas catalysée par la protéine Cas10 mais par les sous-unités Csm3 et Cmr4, respectivement. Ainsi, le système III-A peut dégrader à la fois l’ADN et l’ARN ; on suppose que l’activité de dégradation de l’ARN bien décrite des systèmes III-B est complétée par la capacité de dégrader l’ADN.
Étant donné que les systèmes de type III ne nécessitent pas la présence de PAM dans les cibles, dans leur cas, il doit exister un mécanisme différent de celui des systèmes de type I pour faire la distinction entre l’ADNdb propre et étranger. Dans le cas du complexe Csm, le crRNA est complémentaire non seulement de l’espaceur CRISPR, mais également de la répétition adjacente. Ainsi, lors de la liaison à la molécule cible, l’ARNr se liera uniquement au protoespaceur, et lors de la liaison à l’ADN cellulaire, il se liera également à la répétition voisine, sur la base de laquelle le système III pourra distinguer l’ADN cellulaire de l’ADN étranger. Fait intéressant, dans les systèmes de type III, l’ADN est coupé très près des sites où les bases correspondantes de l’ARNcr et de l’ADN cible ne sont pas appariées. On ne sait pratiquement rien des mécanismes par lesquels de nouveaux espaceurs sont acquis dans les systèmes de type III.
En plus des sous-types III-A et III-B communément reconnus, il a été proposé en 2015 de distinguer également les sous-types III-C et III-D, présents dans certaines archées. Dans les systèmes de type III-C, la protéine Cas10 présente une inactivation du domaine cyclase; de plus, sa séquence d’acides aminés diffère significativement de celle des systèmes Cas10 III-A et III-B. Dans les systèmes III-D, Cas10 n’a pas de domaine HD ; de plus, il existe un gène csx10 unique similaire à cas5. Les systèmes III-C et III-D ne possèdent pas les gènes cas1 et cas2.
En février 2016, des informations sont apparues selon lesquelles chez certaines bactéries dotées de systèmes CRISPR-Cas de type III (par exemple, la bactérie marine Marinomonas mediterranea), au lieu de la protéine Cas1 habituelle, une protéine chimérique Cas1-RT réticulée avec des fonctions de transcriptase inverse. En raison de la présence d’une telle protéine, une bactérie peut intégrer dans son génome des espaceurs formés de génomes pathogènes avec des génomes d’ARN par transcription inverse. Il a été constaté que les systèmes de type III, en particulier Cas10, produisent des seconds messagers oligoadénylés cycliques, convertissant l’ATP en un produit cyclique qui active allostériquement Csm6, qui aide ensuite à dégrader l’ARN viral.
Systèmes de type II
Les systèmes CRISPR-Cas de type II se distinguent en raison de leur base génétique et de leurs mécanismes moléculaires inhabituels. En particulier, les complexes multiprotéiques qui traitent l’ARNcr dans les systèmes de type I et III sont remplacés dans les systèmes de type II par une seule protéine, Cas9, impliquée dans les trois étapes fondamentales de ce système. Ainsi, les systèmes de type II sont le type le plus simple de système CRISPR-Cas. De plus, des éléments supplémentaires propres aux systèmes de type II sont impliqués dans la biogenèse des ARNcr. Les systèmes de type II ne se trouvent que chez les bactéries et parmi les systèmes de types I, II et III, ils sont les moins courants. Cependant, ce sont les systèmes de type II qui ont trouvé une application comme outil d’édition des génomes.
Les systèmes de type II sont divisés en trois sous-types en fonction de la présence et des séquences des gènes cas associés : II-A, II-B et II-C. En plus des gènes cas1 et cas2, communs à tous les systèmes de types I à III, les systèmes de type II possèdent un gène cas9 supplémentaire, qui code pour l’endonucléase Cas9. Cas9 est impliqué dans l’acquisition de nouveaux espaceurs, l’accumulation d’ARNcr et les interférences. De plus, les systèmes II-A contiennent le gène csn2, dont le produit protéique est impliqué dans l’acquisition des espaceurs. Dans les systèmes II-B, ce gène est remplacé par le gène cas4, et les systèmes II-C n’ont ni csn2 ni cas4. La longueur de Cas9 varie selon les sous-types, les systèmes II-C ayant généralement les orthologues les plus courts. La partie centrale de Cas9, qui comprend le domaine nucléase et le cluster riche en arginine caractéristique de cette protéine, est très probablement codée par des gènes dérivés d’éléments génétiques mobiles qui ne sont en aucun cas liés à CRISPR. Compte tenu de la similitude significative des séquences d’acides aminés entre Cas9 et ses homologues, qui ne sont pas associés aux systèmes CRISPR-Cas, Cas9 ne peut être considéré au sens plein du sens de protéine signature des systèmes de type II. Cependant, cela peut être considéré comme une caractéristique de ces systèmes.
La biogenèse des ARNcr dans les systèmes de type II présente un certain nombre de caractéristiques uniques. En particulier, cela nécessite un traitement par la RNase III et la liaison d’ARN CRISPR trans-codés spécifiques (tracrRNA) aux pré-crRNA. Le tracrRNA contient une région complémentaire à la région du crRNA qui a été transcrite à partir de la répétition CRISPR. Au cours du traitement des ARNcr, le tracrARN se lie aux ARNcr qui n’ont pas encore été excisés dans le pré-ARNcr, entraînant la formation d’ARNcr matures. Le complexe crRNA-tracrRNA-Cas9 mature résultant contient un court crRNA dans lequel 20 à 24 nucléotides sont complémentaires de l’extrémité 3′ de l’espaceur et 20 à 24 nucléotides sont complémentaires de l’extrémité 5′ de la répétition. La première étape du traitement des pré-ARNcr se produit dans des régions complémentaires aux répétitions CRISPR ; en conséquence, l’extrémité 3′ du crRNA est formée. La troncature ultérieure de l’extrémité 5′ par des nucléases inconnues se produit dans les séquences correspondant aux espaceurs CRISPR. L’accumulation d’ARNcr dans les cellules nécessite la protéine Cas9, bien que l’on ne sache pas si cela est dû à l’implication de Cas9 dans le traitement de l’ARNcr, à la stabilisation post-traitement de l’ARNcr par Cas9, ou aux deux.
Le complexe crRNA-tracrRNA-Cas9 reconnaît les ADN cibles complémentaires du crRNA et contenant du PAM. Comme dans les systèmes de type I, l’absence de PAM dans les loci CRISPR empêche la coupure de l’ADN cellulaire. Tout d’abord, Cas9 reconnaît le PAM, puis la complémentarité des ARNcr de l’ADN adjacent est vérifiée. La coupe de l’ADN cible est réalisée en introduisant deux cassures simple brin avec les motifs RuvC et HNH de la protéine Cas9, ce qui entraîne la formation d’une cassure double brin aux extrémités franches à l’extrémité du protoespaceur dans le R-boucle la plus proche du PAM, trois nucléotides avant le PAM.
Dans les systèmes III-C (en particulier dans le système Neisseria meningitidis CRISPR-Cas), un mécanisme alternatif de biogenèse des ARNcr a été décrit qui utilise des promoteurs situés dans les répétitions CRISPR. Une direction alternative de transcription peut se produire même sans la participation de la RNase III.
Autres fonctions des systèmes CRISPR-Cas
Malgré le fait que les fonctions des systèmes CRISPR-Cas sont généralement associées à l’immunité adaptative des procaryotes, il existe de nombreuses preuves de la participation de ces systèmes à des processus complètement différents qui ne sont pas liés à la protection contre les éléments génétiques étrangers (par exemple, dans la régulation du comportement de groupe, de la virulence, de la réparation de l’ADN et de l’évolution du génome).
Un exemple est le système CRISPR-Cas chez la delta-protéobactérie prédatrice Myxococcus xanthus, omniprésente dans le sol. Son cycle de vie comprend les étapes de formation de la fructification et de sporulation, au cours desquelles les cellules individuelles s’agrègent et se différencient en myxospores, formant une fructification. En se séparant, les myxospores se transforment en cellules bactériennes individuelles, et ce processus est étroitement régulé par des signaux de détection de quorum et des cascades de signalisation intracellulaires. Le système CRISPR-Cas de cette bactérie appartient au type I-C et comprend 7 gènes Cas et le locus CRISPR contenant 22 espaceurs. En cas de manque de nutriments, le système déclenche la synthèse dans les cellules du signal A, constitué d’acides aminés et de peptides, qui active la transcription du gène fruA (l’opéron cas peut également activer ce gène via la protéine Cas8c). Lorsque les cellules entrent en contact les unes avec les autres, elles forment un signal C codé par le gène csgA, qui active également fruA, qui favorise ensuite l’expression des gènes cas. Ainsi, les gènes cas font partie de la boucle de rétroaction positive avec le gène fruA et sont impliqués dans la formation de la fructification et la sporulation de la bactérie.
Les systèmes CRISPR-Cas pourraient être impliqués dans la régulation de la virulence des bactéries pathogènes. Par exemple, Francisella novicida possède un système de type II composé de quatre gènes cas et d’un locus CRISPR inversé contenant 13 espaceurs. Il régule négativement l’expression de la lipoprotéine bactérienne (BLP), un facteur de virulence de surface. C’est ce dernier qui est reconnu par les récepteurs Toll-like 2 du système immunitaire de l’hôte, une régulation négative du BLP est donc nécessaire pour une infection réussie. On suppose que le complexe de Cas9, du petit ARNcr (scaRNA) et du tracrARN se lie au transcrit blp et le détruit par un mécanisme inconnu. Les systèmes CRISPR-Cas sont impliqués dans la régulation de la virulence chez des bactéries telles que Campylobacter jejuni, Neisseria meningitidis, Legionella pneumophila (dans le cas de cette bactérie, seul cas2 parmi tous les gènes cas est impliqué dans la régulation de la virulence) et Listeria monocytogenes.
Chez de nombreuses bactéries, les systèmes CRISPR-Cas sont utilisés pour réguler leurs propres gènes non liés à la virulence. En particulier, chez Pseudomonas aeruginosa, le système de type I-F est impliqué dans la régulation des gènes associés à la formation du biofilm. En outre, certains suggèrent que les protéines Cas1 et Cas2 peuvent fournir une protection contre les bactériophages, agissant de la même manière que les systèmes toxine-antitoxine, c’est-à-dire provoquant le repos et la mort ultérieure des cellules infectées. Il existe des preuves de l’implication des systèmes CRISPR-Cas dans la réparation de l’ADN. Par exemple, Cas1, qui fait partie du système E. coli de type I – E, peut interagir physiquement avec les enzymes de réparation et de recombinaison. La suppression du gène cas1 ou des loci CRISPR associés a entraîné une sensibilité accrue aux agents endommageant l’ADN et des perturbations dans la ségrégation des chromosomes au cours de la division.
Les systèmes CRISPR-Cas ciblant le chromosome bactérien peuvent jouer un rôle important dans les réarrangements génomiques des bactéries et fournir la base génétique de l’évolution, malgré le fait que dans la plupart des cas, les protéines Cas auto-ciblées conduisent à la mort cellulaire. Dans la bactérie Pectobacterium atrosepticum, il a généralement été démontré que les ARNcr ciblant les îlots chromosomiques acquis par transfert horizontal de gènes entraînaient la mort cellulaire, mais certaines cellules survivantes présentaient d’importantes délétions chromosomiques, notamment la suppression complète d’un îlot cible d’environ 100 paires de bases. Dans ces rares cas, les suppressions ont augmenté la condition physique globale des mutants.
Il est intéressant de noter que les systèmes CRISPR-Cas sont présents non seulement chez les procaryotes, mais également chez les bactériophages et un certain nombre d’autres éléments génétiques mobiles (MGE). Cette circonstance est peut-être associée à la propagation des systèmes CRISPR-Cas chez les bactéries et les archées par transfert horizontal de gènes. Les systèmes CRISPR-Cas de ces éléments peuvent être ciblés sur d’autres FEM, fournissant ainsi des mécanismes de concurrence entre les FEM. Les MGE portant les systèmes CRISPR-Cas peuvent rivaliser avec les îlots de pathogénicité bactérienne qui sont excisés du génome lors de l’infection par les phages et transférés à d’autres bactéries dans les capsides des phages. En utilisant les capsides des phages pour leur propre transmission, les îlots de pathogénicité peuvent bloquer complètement la reproduction des phages. Un exemple est le système CRISPR-Cas du phage ICP1 Vibrio cholerae, qui appartient au type I-F et possède 2 gènes cas et 9 espaceurs (apparemment, il est homologue du système Yersinia pestis). L’un des espaceurs est complémentaire de l’îlot de pathogénicité de Vibrio cholerae, de sorte que le phage peut rivaliser avec les îlots de pathogénicité pour les capsides. De plus, le système CRISPR-Cas ICP1 peut acquérir de nouveaux espaceurs, ce qui permet au phage de co-évoluer avec la bactérie hôte.
En 2016, des informations sont apparues selon lesquelles les grands virus nucléaires-cytoplasmiques contenant de l’ADN possèdent un système de protection ressemblant à CRISPR et conçu pour protéger contre les virophages (en particulier le virophage Zamilon dans Mimivirus). Ce système de protection a été nommé MIMIVIRE.
Anti-CRISPR
Il a été constaté qu’en réponse à la propagation de certains espaceurs CRISPR dans la population bactérienne (et, par conséquent, à la propagation de la résistance aux bactériophages correspondants), les bactériophages mutent intensément et perdent même les parties du génome qui servent le plus souvent de cibles aux systèmes CRISPR-Cas et s’intègrent dans le génome bactérien en tant qu’espaceurs
Certains phages codent pour des protéines spécifiques (protéines anti-CRISPR, Acr) qui interfèrent avec les systèmes CRISPR-Cas et favorisent l’infection. L’analyse des phages de Pseudomonas aeruginosa a permis d’isoler plusieurs variétés de protéines Acr. Initialement, les protéines Acr ont été décrites dans des souches de P. aeruginosa portant des prophages dans leurs chromosomes. Bien que la plupart de ces souches aient un système CRISPR-Cas de type IF actif, chez certaines souches, le système est resté inactif même en présence d’espaceurs ciblant les phages. L’analyse moléculaire des souches avec des systèmes inactifs a révélé un certain nombre de petites protéines codées par les phages qui étaient responsables du développement du phénotype sensible aux phages. Les protéines Acr peuvent supprimer le fonctionnement des systèmes CRISPR-Cas de diverses manières, notamment (dans le cas des systèmes de type I-F) en se liant au complexe Cascade et en bloquant sa liaison à l’ADN cible ou en se liant aux protéines Cas, conduisant à la perte de leur activité nucléasique.
On connaît la protéine Acr, qui empêche la liaison de l’hélicase nucléase Cas3 au complexe d’ARNcr et d’autres protéines Cas déjà liées à son ADN cible. Puisque le complexe de Cas et d’ARNcr associé à l’ADN ne permet pas à l’appareil de transcription de se lier à l’ADN, cette protéine Acr transforme le complexe d’ARNcr et de Cas en un répresseur transcriptionnel. Il s’agit d’une régulation de l’activité du système CRISPR-Cas par un facteur protéique. Les protéines Acr peuvent présenter une forte spécificité pour le système CRISPR-Cas ; en particulier, les protéines qui bloquaient le système I-F de P. aeruginosa n’avaient aucun effet sur le système I-F de P. aeruginosa ou d’E. coli. Cependant, certains phages qui possèdent des gènes suppresseurs pour le système I-F de P. aeruginosa codent également pour de petites protéines suppressives qui suppriment le système I-E de P. aeruginosa, mais pas le système I-E d’E. coli.
L’émergence de mécanismes de protection contre les interférences CRISPR dans les phages est considérée comme le résultat d’une longue coévolution des phages et de leurs hôtes.
Signification évolutive des systèmes CRISPR-Cas
Selon Eugene V. Koonin (Logic of Chance: The Nature and Origin of Biological Evolution, 2014), le fonctionnement des systèmes CRISPR-Cas peut être considéré comme un processus évolutif qui satisfait au scénario évolutif de Lamarck, à savoir les critères suivants: (1) les changements génomiques dans les locus CRISPR (insertion de nouveaux espaceurs) sont provoqués par l’influence de l’environnement (plus précisément, des éléments génétiques étrangers), (2) les changements sont limités à des loci génomiques spécifiques, (3) les changements permettent une adaptation à un impact spécifique (à un élément génétique étranger spécifique).
Cependant, cette vision de CRISPR a été critiquée. Selon A. Wyss, en effet, la correspondance de CRISPR-Cas aux critères lamarckiens n’est que superficielle. Les systèmes CRISPR-Cas présentent certaines des propriétés de l’évolution darwinienne, en particulier l’acquisition apparemment aléatoire d’espaceurs au niveau de la population, suivie de la sélection des clones survivants présentant la meilleure forme physique.
Identification des systèmes CRISPR-Cas
Les systèmes CRISPR-Cas sont répandus parmi les bactéries et les archées, et leur caractéristique est l’alternance de séquences répétitives et d’espaceurs. En raison de cette caractéristique, les loci CRIPSR sont assez faciles à trouver dans de longues séquences d’ADN, car avec l’augmentation du nombre de répétitions dans un locus, la probabilité d’un résultat faussement positif diminue. Parmi les programmes utilisés pour rechercher CRISPR en fonction de la recherche de répétitions séparées par des espaces dans de longues séquences figurent CRT, PILER-CR et CRISPRfinder. Trouver CRISPR dans les données métagénomiques est plus difficile: en utilisant des algorithmes standards, les locus CRISPR ne peuvent pas être collectés en raison de la présence de nombreuses répétitions, ainsi que de variations spécifiques à la souche. La réaction en chaîne par polymérase peut être utilisée pour augmenter le nombre de locus CRISPR, puis analyser le contenu des espaceurs, cependant, cette méthode ne fournit que des informations sur un locus CRISPR spécifique et n’est applicable qu’aux organismes (par exemple Streptococcus ; Sulfolobus islandicus) dont le génome est disponible dans des bases de données (afin que des amorces appropriées puissent être générées).
Application de CRISPR en génie génétique
Avant la découverte des fonctions et des mécanismes d’action des systèmes CRISPR-Cas en tant que méthodes d’édition du génome spécifique à un locus, des méthodes basées sur l’utilisation de nucléases à doigt de zinc (Zinc-finger nucleases ou ZFN), ainsi en tant qu’endonucléases TAL, ont été développées de manière plus intensive (Transcription activator-like effector nuclease, TALEN). Ces méthodes sont plutôt laborieuses, peu efficaces et coûteuses : pour chaque nouveau locus cible, le développement, l’expression et la vérification d’une toute nouvelle paire de polypeptides sont nécessaires, ce qui limite considérablement la portée de ces méthodes.
Cependant, en 2012-2013, des méthodes fondamentalement nouvelles de manipulation du matériel génétique basées sur l’utilisation des systèmes CRISPR-Cas sont apparues en génie génétique. Ces méthodes conviennent à l’édition ciblée des génomes des procaryotes et des eucaryotes (bien que ces derniers ne disposent pas de leur propre système CRISPR-Cas, il s’est toutefois avéré que des éléments du système CRISPR-Cas d’origine bactérienne introduits artificiellement dans un eucaryote la cellule est capable de fonctionner dans un nouvel environnement). Dans le même temps, les technologies CRISPR-Cas modernes utilisent la protéine Cas9, qui est la même pour tous les loci cibles, et la spécificité d’action n’est pas déterminée par la protéine, mais par l’ARNcr. Les méthodes basées sur ZFN et TALEN sont encore utilisées aujourd’hui et sont même préférées pour la recherche clinique, cependant, la simplicité, l’efficacité et la rentabilité des méthodes utilisant le système CRISPR-Cas9 en ont fait un choix de premier ordre pour l’édition ciblée du génome et la liaison à l’ADN.
Les méthodes basées sur CRISPR-Cas9 sont proches des mécanismes d’action naturels de ces systèmes: l’ARN est utilisé pour reconnaître une séquence cible située à proximité du PAM, et la nucléase Cas9 qu’il dirige produit une cassure double brin au niveau du site cible. Cependant, lors de l’édition du génome eucaryote, le résultat du travail de CRISPR-Cas9 n’est pas la destruction de la molécule d’ADN entière, mais la réparation de la cassure double brin produite par Cas9. La réparation peut être réalisée à la fois par assemblage d’extrémités non homologues (NHEJ) et par recombinaison homologue. La réparation par jonction d’extrémité non homologue entraîne souvent de petites insertions ou délétions qui peuvent perturber le cadre de lecture des gènes codant pour les protéines, entraînant une perte de fonction du gène cible. En provoquant de nombreuses cassures double brin, des délétions importantes, voire des inversions, peuvent être obtenues.
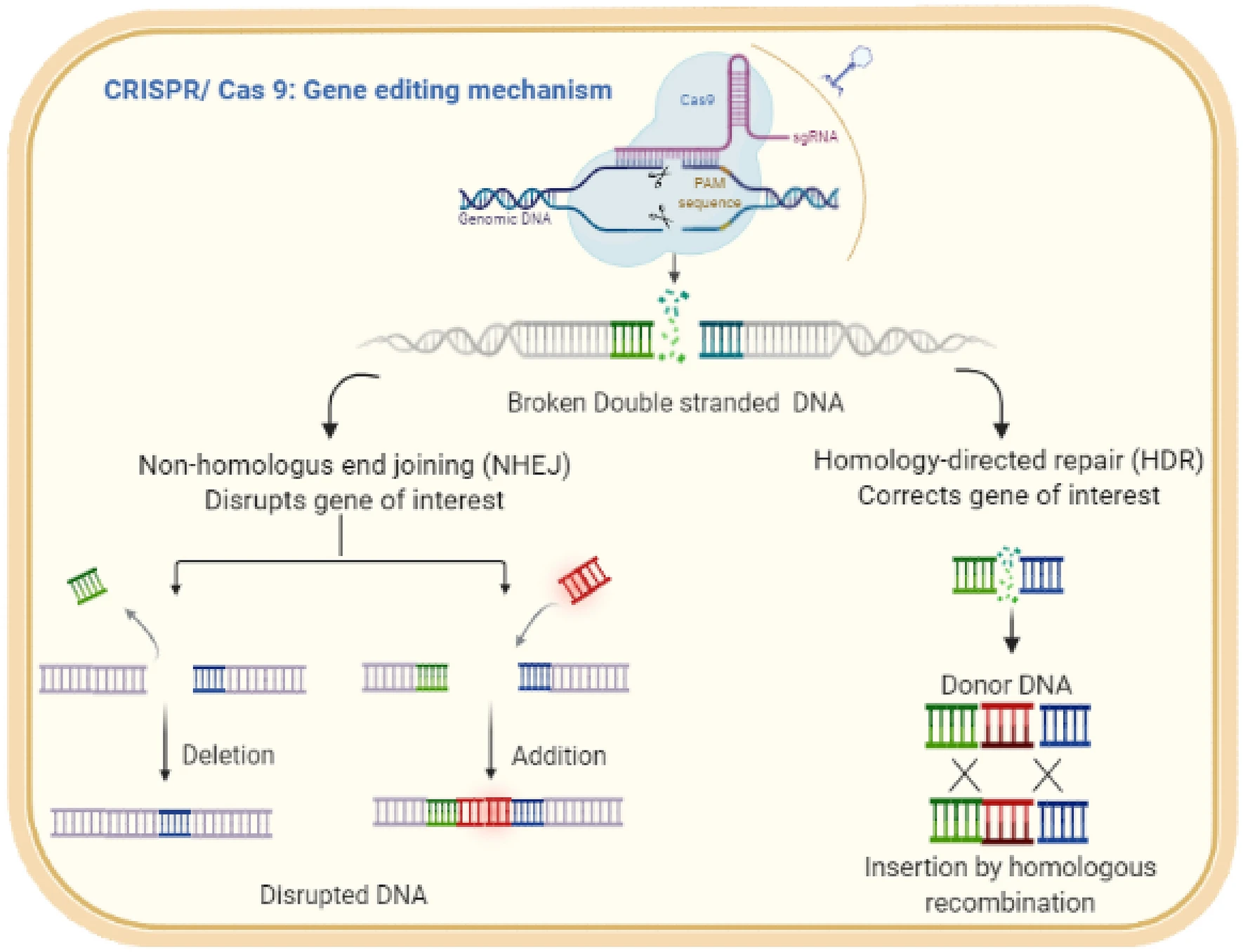
En revanche, la réparation par recombinaison homologue consiste à remplacer la séquence supprimée par une nouvelle séquence complémentaire du modèle de réparation créé par le chercheur lui-même. Ainsi, la recombinaison homologue peut être utilisée pour supprimer les mutations indésirables, créer de nouveaux allèles, insérer ou fusionner des domaines fonctionnels. De plus, l’inactivation mutationnelle des domaines RuvC ou HNH Cas9 convertit cette protéine en une nickase dirigée par l’ARN produisant des cassures simple brin plutôt que des cassures double brin. L’inactivation des deux domaines convertit Cas9 en une protéine de liaison à l’ADN guidée par l’ARN qui ne coupe pas la cible. Dans ce cas, un domaine avec d’autres fonctions peut être attaché au domaine de liaison à l’ADN, ce qui, à son tour, peut provoquer divers changements dans le locus cible: activation ou répression de la transcription, modification de la chromatine, formation accrue de boucles et bien d’autres. De plus, la forme inactivée de Cas9 (dCas9, Cas9 «mort») sert de base à de nouvelles techniques de recherche, telles que l’imagerie par fluorescence ou la création d’étiquettes pour l’isolement physique ultérieur des locus.
Malgré l’efficacité de l’utilisation des systèmes CRISPR-Cas, l’origine de Cas9 impose certaines restrictions sur le choix des cibles ADN: par exemple, lorsqu’on utilise Streptococcus pyogenes Cas9, seules les séquences suivies par PAM, à savoir 5′-NGG (où N est n’importe quel nucléotide). Cependant, la nécessité du PAM n’impose pas de restrictions sérieuses à l’utilisation des systèmes CRISPR-Cas9: dans le génome humain, de telles séquences apparaissent presque tous les 8 à 12 nucléotides. Avant d’être utilisé dans des constructions génétiques, le gène Cas9 doit être préalablement optimisé pour les codons utilisés en fonction de l’organisme dont le génome est censé être modifié: le gène cas9 de S. pyogenes a une faible teneur en GC (35%), et pour les organismes dont le génome a une composition GC élevée, une optimisation des codons Cas9 peut être nécessaire.
Actuellement, le système de type CRISPR-Cas II est utilisé pour l’édition du génome, et la protéine SpyCas9 (nucléase Cas9 de la bactérie S. pyogenes) est le plus souvent utilisée ; cependant, des protéines Cas9 alternatives sont en cours de développement, ce qui augmentera la portée de CRISPR-Cas. Par exemple, les formes tronquées de Cas9 peuvent reconnaître diverses séquences PAM. Bien que l’édition du génome puisse être réalisée efficacement avec le crRNA et le tracrRNA transcrits séparément, le développement de la technologie de l’ARN guide unique (sgRNA) a simplifié ce système. Dans ce cas, le système à quatre composants RNase III:crRNA:tracrRNA:Cas9 est remplacé par le système à deux composants sgRNA:Cas9. Actuellement, le sgRNA est utilisé beaucoup plus fréquemment que les crRNA et tracrRNA séparés. Enfin, des développements sont en cours pour améliorer la spécificité de Cas9 et réduire les effets secondaires. Début 2016 ont été publiés les résultats des travaux de chercheurs américains, qui ont réussi à réduire le nombre d’erreurs à presque zéro.
La délivrance de sgRNA et de Cas9 aux cellules cibles est assurée par diverses méthodes. Par exemple, des plasmides codant pour sgRNA et Cas9 peuvent être utilisés à cet effet, et les cellules peuvent être transfectées (ou transformées, dans le cas des procaryotes) avec eux. De tels plasmides peuvent être introduits dans les cellules par électroporation. Dans certains cas, il est plus pratique d’utiliser des plasmides codant pour Cas9 et de délivrer l’ARN sous forme d’amplicons générés par PCR.
En 2015, une nouvelle méthode a été proposée pour introduire le sgRNA et le Cas9 dans la cellule à l’intérieur de nanobobines spéciales. Une telle nanobobine est constituée d’une chaîne d’ADN densément entrelacée, dont l’une des sections est complémentaire du sgARN transféré ; ainsi, le complexe sgRNA:Cas9 est fixé à l’intérieur de la bobine. De plus, la nanobobine est capable de s’auto-assembler. De nombreux complexes sgRNA:Cas9 différents peuvent être attachés à une seule nanobobine. Au contact de la cellule, la nanobobine pénètre dans l’endosome, mais un polymère spécial recouvrant la nanobobine assure la destruction de l’endosome et permet au sgRNA:Cas9 d’atteindre le noyau.
Modifications de la méthode
Pour l’édition ciblée du génome des cellules eucaryotes, on utilise non seulement le Cas9 de S. pyogenes, mais aussi le Cas9 de Streptococcus thermophilus, Neisseria meningitidis, ainsi que Cas9 de Staphylococcus aureus (SaCas9), qui est 25% plus petit que SpyCas9, ce qui lui permet d’être conditionné dans un virus adéno-associé (AAV) pour l’administration du vecteur dans les cellules d’un organisme vivant en tant qu’agent thérapeutique.
Une forme de Cas9 (dCas9) incapable de couper l’ADN a trouvé de nombreuses applications. L’utilisation de dCas9 réticulé à une protéine fluorescente est à la base de la nouvelle méthode CASFISH (CRISPR-Cas9 mediated fluorescence in situ hybridization), qui permet le marquage fluorescent des loci cibles. Grâce à ce dCas9, on peut suivre la longueur des télomères, ainsi qu’observer la dynamique de certains loci au cours du cycle cellulaire.
La forme dCas9 peut être utilisée pour supprimer la transcription d’un gène cible (lorsqu’elle se lie à ce dernier au niveau du promoteur, des régions régulatrices ou du début de la région codante) ; de plus, un peptide répresseur peut être ligaturé à dCas9 pour supprimer la transcription. Au contraire, dCas9 réticulé avec des protéines activant la transcription (facteurs de transcription et effecteurs) peut activer la transcription du gène cible. De plus, des endonucléases de restriction artificielles, ainsi que des enzymes qui modifient l’épigénome (ADN méthyltransférases, histones acétyltransférases) et régulent ainsi l’activité des gènes cibles, peuvent être attachées à dCas9. En 2016, des cellules souches embryonnaires de souris ont été reprogrammées en deux lignées extra-embryonnaires (cellules trophoblastiques et endodermiques extra-embryonnaires) en activant les gènes Cdx2 et Gata6 à l’aide d’activateurs médiés par CRISPR.
De plus, dCas9 peut être lié à un monomère de l’endonucléase FokI, qui fonctionne comme des dimères. Les dimères FokI peuvent introduire des cassures double brin dans les séquences cibles. Deux sgRNA sont utilisés pour diriger dCas9 réticulé au monomère FokI, ce qui augmente considérablement la précision du système. Lorsque deux monomères, chacun guidé par son propre sgRNA, sont distants d’environ 30 pb, FokI se dimérise et introduit une cassure double brin. Pour éliminer les locus associés au sgRNA, dCas9 portant certains épitopes peut être utilisé. En fait, cette méthode est une variante particulière de l’immunoprécipitation de la chromatine.
Des analogues de Cas9 ont été découverts et peuvent cliver des molécules d’ARN au lieu de l’ADN. L’utilisation de ces protéines permettra d’éditer ou de supprimer sélectivement l’activité des miARN. Francisella novicida Cas9 (FnCas9) peut être reprogrammée pour cibler le génome de l’ARN du virus de l’hépatite C, entraînant la suppression du cycle de vie viral dans les cellules eucaryotes. Sur la base de ce système, des centaines de remèdes contre divers virus peuvent être créés.
À l’automne 2015, une nouvelle méthode a été proposée, alternative à CRISPR-Cas9 – CRISPR-Cpf1. Cpf1 est une endonucléase qui est une protéine effectrice des systèmes CRISPR-Cas de type V. Il est plus petit que Cas9 et ne nécessite que du crRNA pour fonctionner, pas du tracrRNA. À cet égard, il est possible que dans certains cas, la méthode CRISPR-Cpf1 soit plus pratique que la méthode CRISPR-Cas9.
En 2015, une nouvelle méthode d’autoclonage CRISPR (self-clonage CRISPR) a également été proposée. Dans ce cas, un plasmide contenant un sgARN palindromique autoclonant est introduit dans les cellules, ainsi qu’un court ADN double brin contenant la séquence codant pour le sgARN souhaité. Lorsque le plasmide est transcrit, le sgARN résultant complexé avec Cas9 se lie de manière complémentaire à la séquence du plasmide codant pour le sgARN. Cas9 introduit une cassure double brin qui est réparée par recombinaison homologue en utilisant l’ADN double brin introduit comme matrice ; en conséquence, le plasmide contient à nouveau la séquence codant pour le sgARN souhaité. Contrairement à la méthode CRISPR standard, qui nécessite une production longue et laborieuse de plasmides spéciaux pour chaque nouveau locus cible, la méthode CRISPR d’autoclonage peut réduire la durée de l’expérience de six jours à trois heures et réduire son coût de six fois.
Actuellement, des méthodes chimiques sont intensivement développées pour contrôler le fonctionnement de CRISPR-Cas: dose, durée d’action, spécificité et autres paramètres.
Importance biotechnologique et médicale
Actuellement, les méthodes CRISPR-Cas sont utilisées avec succès dans le génie génétique de divers organismes : eucaryotes multicellulaires et unicellulaires (levure), et procaryotes. L’utilisation de CRISPR-Cas chez les micro-organismes permet de modifier leurs voies métaboliques, ce qui ouvre des opportunités pour le développement de nouvelles stratégies biotechnologiques. De plus, la création de souches de bactéries technologiquement importantes et résistantes à divers phages grâce à CRISPR-Cas revêt une grande importance pour la biotechnologie.
Des méthodes d’édition des génomes à l’aide de CRISPR-Cas ont été développées pour des organismes modèles (par exemple, des souris, la mouche des fruits Drosophila melanogaster, le nématode Caenorhabditis elegans, le poisson zèbre, et d’autres). De telles méthodes ont été utilisées pour modifier le génome de champignons, en particulier le champignon filamenteux Aspergillus oryzae, utilisé dans l’industrie pour la fermentation du soja et du champignon. L’édition CRISPR-Cas de cultures cellulaires de mammifères, y compris humaines, est d’une grande importance. En 2017, le génome d’embryons humains a été édité par cette méthode.
Des travaux sont en cours pour modifier les génomes à l’aide de CRISPR-Cas chez les bovins, les porcs et d’autres animaux de grande importance économique, comme les abeilles. En novembre 2015, les résultats d’une expérience dans laquelle 62 rétrovirus endogènes ont été inactivés dans le génome porcin à l’aide de la technologie CRISPR-Cas ont été publiés. Les auteurs de l’étude espèrent que ces résultats rendront possible à l’avenir la xénotransplantation d’organes de porc à l’homme. Enfin, la mutagenèse CRISPR-Cas peut être utilisée pour contrôler les espèces envahissantes (par exemple, la mouche envahissante Drosophila suzukii).
La technologie CRISPR-Cas a été appliquée avec succès au génie génétique des plantes, y compris des plantes ornementales (par exemple, les pétunias) et de nombreuses cultures agricoles importantes : le riz, le soja, le blé, le sorgho, du maïs, de la tomate et une orange. La possibilité d’introduire des systèmes CRISPR-Cas dans des plantes cultivées pour créer une immunité antivirale est à l’étude. Le système CRISPR-Cpf1 peut également être utilisé pour le génie génétique végétal.
Les méthodes basées sur CRISPR-Cas peuvent également être utilisées en médecine pour le traitement d’une grande variété de maladies : virales (y compris l’infection par le VIH, et les infections à herpèsvirus), les allergies et les maladies immunologiques (y compris auto-immunes, oncologiques, les maladies cardiovasculaires, voire les rhumatismes, ainsi que les troubles héréditaires tels que le syndrome de Down, la drépanocytose, la rétinite pigmentaire et la β-thalassémie. En 2013, une publication rapportait que des chercheurs avaient réussi à modifier un gène anormal dans les cellules souches d’un patient atteint de mucoviscidose. Il est possible que le système CRISPR-Cas puisse aider au traitement de la dystrophie musculaire de Duchenne (DMD): il a été démontré que CRISPR-Cas peut restaurer le gène de la dystrophine dans une culture cellulaire DMD.
On suppose que de telles cellules dotées d’un génome «réparé» peuvent être transplantées dans le corps du patient, où elles peuvent remplacer les cellules malades et remplir les fonctions nécessaires. En octobre 2016, une édition CRISPR/Cas du génome humain adulte a été réalisée en Chine: un patient atteint d’un cancer du poumon a reçu une injection de lymphocytes T modifiés par CRISPR-Cas. Les chercheurs pensent que la modification du génome du moustique porteur du paludisme avec CRISPR-Cas peut aider à lutter contre le paludisme. La possibilité de modifier le génome d’un autre protozoaire pathogène important, Toxoplasma gondii, à l’aide de CRISPR-Cas a été démontrée.
Le système CRISPR-Cas peut être utilisé pour obtenir des tissus résistants à l’inflammation à partir de cellules pluripotentes humaines. Les méthodes CRISPR-Cas se sont révélées efficaces pour manipuler le locus PRPN, qui code pour une protéine prion responsable d’un certain nombre de maladies neurodégénératives chez l’homme et d’autres mammifères. Les lignées cellulaires modifiées avec CRISPR-Cas peuvent être utilisées comme modèles pour diverses maladies humaines. Par exemple, en utilisant CRISPR-Cas, des cellules présentant des mutations correspondant à deux maladies rénales (maladie polykystique des reins et glomérulosclérose segmentaire focale) ont été obtenues à partir d’une lignée cellulaire pluripotente humaine. Plus tard, des mini-organes correspondant aux reins d’une personne atteinte de ces maladies ont été développés à partir de ces cellules. La même méthode a été utilisée pour modéliser le syndrome du QT long sur les cardiomyocytes. De tels modèles peuvent contribuer à l’étude des maladies et au développement de nouveaux médicaments.
Modification de l’ADN pour lutter contre l’infection par le VIH
En novembre 2018, il est devenu connu qu’une équipe de scientifiques chinois dirigée par He Jiankui avait réussi à créer les premières personnes au monde avec des gènes artificiellement modifiés (CCR5 désactivé) – deux jumelles censées être immunisées contre le virus de l’immunodéficience humaine (VIH). L’expérience a été critiquée pour avoir violé de nombreuses règles scientifiques et éthiques.
Réaction du public
En 2015, au moins quatre laboratoires aux États-Unis, en Chine et au Royaume-Uni, ainsi que la société américaine de biotechnologie Ovascience, ont annoncé leur intention de modifier le génome des embryons humains à l’aide de CRISPR-Cas. À la lumière de ces développements, de nombreux scientifiques ont préconisé un moratoire international sur l’utilisation de la technologie CRISPR-Cas dans les embryons humains et les cellules germinales, y compris à des fins médicales. Ces scientifiques ont soutenu la poursuite des recherches fondamentales sur CRISPR, cependant, à leur avis, la technologie CRISPR-Cas n’est pas encore suffisamment développée pour garantir l’absence de mutations indésirables et de défauts héréditaires chez les patients lorsqu’elle est appliquée dans la pratique clinique.
En avril 2015, un groupe de scientifiques chinois a publié un article dans la revue Protein & Cell rapportant les résultats de leur tentative de modifier l’ADN d’embryons humains non viables à l’aide de CRISPR-Cas. Ils essayaient de corriger la mutation conduisant à la bêta-thalassémie. Selon le chercheur principal, Nature et Science ont rejeté l’article en raison de considérations éthiques. Les résultats de l’expérience n’étaient pas très optimistes en raison des nombreuses mutations survenues en dehors du gène cible. Les auteurs de l’étude ont déclaré que la technologie CRISPR-Cas n’est pas encore prête à être utilisée en médecine reproductive.
En décembre 2015, le Sommet international sur l’édition des gènes humains s’est tenu à Washington sous la présidence de David Baltimore. Lors de ce sommet, des représentants des académies nationales des sciences des États-Unis, de Grande-Bretagne et de Chine ont discuté des questions éthiques liées à la modification génétique des cellules germinales humaines. Au cours de la réunion, il a été décidé de poursuivre la recherche fondamentale et clinique sur des bases juridiques et éthiques appropriées. Une attention particulière a été attirée sur la différence entre l’utilisation clinique des cellules somatiques, dans lesquelles la propagation des mutations produites est limitée à un seul individu, et les cellules germinales, dont les anomalies génomiques peuvent être héritées par la génération suivante. Ces dernières pourraient avoir des conséquences imprévues et de grande envergure sur l’évolution humaine, tant génétiques que culturelles.
En février 2016, un groupe de scientifiques britanniques a obtenu l’autorisation de modifier génétiquement des embryons humains à l’aide de CRISPR-Cas et de méthodes associées.