{kind=link}
La nécrose (du grec ancien νέκρωσις (nékrōsis) «mort») est une forme de lésion cellulaire qui entraîne la mort prématurée des cellules des tissus vivants par autolyse. La nécrose est causée par des facteurs externes à la cellule ou au tissu, tels qu’une infection ou un traumatisme, qui entraînent la digestion non régulée des composants cellulaires. En revanche, l’apoptose est une cause naturelle programmée et ciblée de mort cellulaire. Alors que l’apoptose procure souvent des effets bénéfiques à l’organisme, la nécrose est presque toujours préjudiciable et peut être mortelle. La mort cellulaire due à la nécrose ne suit pas la voie de transduction du signal apoptotique, mais plutôt divers récepteurs sont activés et entraînent la perte de l’intégrité de la membrane cellulaire et une libération incontrôlée des produits de la mort cellulaire dans l’espace extracellulaire. Ceci initie dans les tissus environnants une réponse inflammatoire, qui attire les leucocytes et les phagocytes voisins qui éliminent les cellules mortes par phagocytose. Cependant, les substances nocives microbiennes libérées par les leucocytes créeraient des dommages collatéraux aux tissus environnants. Cet excès de dommages collatéraux inhibe le processus de guérison. Ainsi, une nécrose non traitée entraîne une accumulation de tissus morts en décomposition et de débris cellulaires au niveau ou à proximité du site de la mort cellulaire. Les signes structurels qui indiquent une lésion cellulaire irréversible et la progression de la nécrose comprennent une agglutination dense et une perturbation progressive du matériel génétique, ainsi qu’une perturbation des membranes des cellules et des organites.
Introduction
Les lésions cellulaires irréversibles et la mort cellulaire éventuelle due à des processus pathologiques sont appelées nécrose. Il s’agit d’une mort cellulaire incontrôlée qui entraîne un gonflement des organites cellulaires, une rupture de la membrane plasmique et une éventuelle lyse de la cellule, ainsi qu’un déversement du contenu intracellulaire dans les tissus environnants, entraînant des lésions tissulaires. Contrairement à la mort cellulaire programmée connue sous le nom d’apoptose qui génère des signaux intrinsèques, la nécrose se produit en raison d’un stimulus nocif écrasant provenant de l’extérieur de la cellule et est presque toujours associée à des réponses inflammatoires dues à la libération de protéines de choc thermique, d’acide urique, d’ATP, d’ADN et de protéines nucléaires, qui provoquent l’activation de l’inflammasome et la sécrétion de la cytokine pro-inflammatoire interleukin-1β (IL-1β ou IL1).
La différence la plus significative entre la mort cellulaire programmée (apoptose et autophagie) et la nécrose est la fuite de la membrane plasmique et l’induction consécutive d’une inflammation dans le tissu affecté causée par la libération de composants intracellulaires.
Causes de la nécrose
La nécrose est toujours considérée comme un processus accidentel dans lequel plusieurs facteurs exercent des effets sur les cellules pour commencer l’événement d’élimination. La mort cellulaire nécrotique contient une grande diversité de processus de mort cellulaire. Les facteurs de stress cellulaire comme le manque d’oxygène (hypoxie), les cytokines, l’ischémie (apport sanguin restreint), la chaleur, l’irradiation, les agents pathogènes et l’exposition aux toxines peuvent tous entraîner une nécrose. Ces stimuli provoquent plusieurs changements au niveau cellulaire. Les lésions cellulaires peuvent aller de lésions externes à des anomalies internes.
Une nécrose peut survenir en cas de dommages importants tels qu’une température élevée et un stress mécanique, entraînant la destruction de l’intégrité cellulaire. Dans ce cas, la nécrose est passive et ne nécessite pas de voies de signalisation spécifiques. Un autre type de nécrose, appelée nécrose secondaire, survient au stade tardif de l’apoptose ou de l’autophagie lorsque les cellules mortes ne peuvent pas être éliminées par phagocytose. La nécrose secondaire est considérée comme indépendante des événements de signal initiaux tels que l’apoptose ou l’autophagie. Cependant, la nécrose peut également être le résultat d’une cascade de signalisation. Les premières études ont montré que le diabète multipliait par quatre la nécrose des cardiomyocytes. Les causes les plus courantes de stimulus préjudiciable comprennent :
⊛ Hypoxie : Cela peut survenir en raison d’une ischémie, d’un choc ou d’une insuffisance respiratoire.
⊛ Agents physiques : Ceux-ci comprennent les blessures externes telles que les traumatismes, les températures extrêmes, l’exposition aux radiations ou les chocs électriques.
⊛ Agents chimiques : il s’agit notamment des poisons, de l’exposition professionnelle, de la toxicité des médicaments ou des drogues récréatives.
⊛ Agents biologiques : bactéries, virus ou champignons.
⊛ Réactions immunologiques : réponses auto-immunes.
Mécanismes physiopathologiques
Alors que les espèces réactives de l’oxygène sont produites par les mitochondries comme un processus normal, dans des conditions pathologiques, les molécules d’oxygène réactif augmentent et induisent des dommages dans les biomolécules, ce qui conduit les cellules vers la nécrose. Au cours de la nécrose, les niveaux d’espèces réactives de l’oxygène et de calcium intracellulaire augmentent. Il est important de considérer que l’environnement cellulaire interne est hautement régulé, de sorte que certains stimuli sont capables de modifier la perméabilité de la membrane cellulaire et ainsi de produire un déséquilibre entre différents ions, tels que le potassium, le sodium et le calcium. Le calcium est régulé par le réticulum endoplasmique et une perte d’homéostasie du calcium peut entraîner plusieurs altérations intracellulaires. En revanche, des niveaux accrus de calcium peuvent affecter diverses fonctions mitochondriales et entraîner des altérations de la production d’espèces réactives de l’oxygène. C’est ce qui arrive dans la nécrose.
En effet, l’Adénosine Triphosphate (ATP) est un nucléotide formé à partir d’un nucléoside associé à un triphosphate et est produite par phosphorylation oxydative dans les mitochondries en présence d’oxygène. L’augmentation du calcium cytosolique et le stress oxydatif entraînent des dommages mitochondriaux. Lorsque des niveaux élevés de calcium sont maintenus au fil du temps, ils perturbent l’intégrité de la membrane interne mitochondriale et entraînent une perte de la capacité à générer de l’ATP. Dans la nécrose associée à l’hypoxie ou à une lésion chimique, il y a un manque d’apport d’oxygène aux cellules, entraînant une diminution de la production d’ATP. Ce manque d’ATP entraîne la défaillance de la pompe à sodium dépendante de l’énergie dans la membrane plasmique, provoquant un afflux de calcium et d’eau, entraînant un gonflement des cellules et un détachement des ribosomes du réticulum endoplasmique.
Le calcium cytosolique peut également conduire à l’activation de plusieurs enzymes cytosoliques, y compris les phospholipases et les protéases, qui conduisent à la dégradation des membranes (y compris les membranes lysosomales) et des protéines. En plus, les taux de calcium cytosolique modifiés peuvent activer différents types de protéases, y compris les calpaïnes. Les calpaïnes sont des protéases à cystéine intracellulaires présentes sous forme inactive, qui peuvent être activées par une augmentation du calcium cytosolique. Une fois activés, ils peuvent perturber la membrane lysosomale avec la libération consécutive de cathepsines B et L. Ce groupe de réactions provoque une déstabilisation du système membranaire final. Ensemble, ces altérations font que la cellule perd ses membranes de sorte que le contenu cellulaire est libéré dans l’espace extracellulaire.
Enfin, la mort nécrotique est presque toujours associée à une réponse inflammatoire. Les cellules nécrotiques libèrent des facteurs tels que la protéine HMGB1 (High Mobility Group Box 1) et le facteur de croissance dérivé de l’hépatome (Hepatoma-derived growth factor ou HDGF – également connu sous le nom de High mobility group protein 1-like 2 ou HMG-1L2). Ces facteurs sont détectés par une protéine réceptrice nod-like 3 (NLRP3), qui est une protéine centrale de l’inflammasome. Cela entraîne l’activation de l’inflammasome et provoque la libération de la cytokine pro-inflammatoire IL1β. L’activation de l’inflammasome NLRP3 est déclenchée principalement par l’ATP produit par les mitochondries libérées par les cellules endommagées.
Anatomopathologie
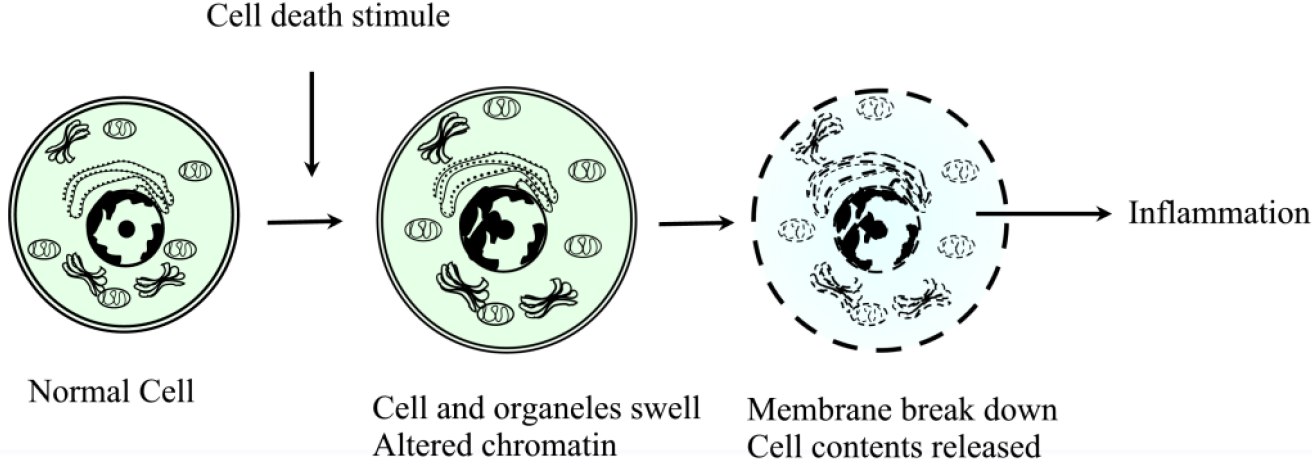
Le mécanisme cellulaire qui conduit à la nécrose est la perte de l’intégrité de la membrane cellulaire à la suite d’une exposition à un stimulus nocif ; cela permet aux ions extracellulaires de se déplacer à l’intérieur de la cellule, suivis d’un liquide conduisant à un éventuel gonflement de la cellule et de ses organites. Un autre mécanisme cellulaire est la perturbation de la membrane lysosomale, qui conduit à la libération d’enzymes protéolytiques dans la cellule, telles que les protéases, la RNAase, les DNAases et les phosphatases. Ceux-ci, lorsqu’ils sont activés dans le cytosol, entraînent des dommages à l’ADN, à l’ARN et aux protéines. Ces enzymes provoquent la digestion des composants cellulaires provoquant la destruction des cellules. Ces deux mécanismes entraînent une perturbation de la membrane plasmique entraînant le déversement du contenu intracellulaire dans les tissus environnants.
Morphologiquement, les cellules nécrotiques se manifestent de manière caractéristique par un gonflement des organites (tels que le réticulum endoplasmique et les mitochondries), la rupture de la membrane plasmique et la lyse de la cellule. Ces changements rendent les cellules plus éosinophiles, vitreuses et vacuolées. La perte de l’intégrité de la membrane cellulaire et la compromission des membranes des organites s’y ajoutent. Globalement, les principales caractéristiques de la nécrose au niveau microscopique sont : Gonflement de la cellule, Gonflement du noyau, Caryolyse (dissolution nucléaire), Karyorrhexis (fragmentation nucléaire), Pycnose nucléaire, Cytoplasme éosinophile pâle, Des vacuoles cytoplasmiques peuvent être présentes dans les zones de nécrose, Des débris cellulaires adjacents et des cellules inflammatoires peuvent être présents en cas de fuite de la membrane cellulaire. Ces différents modèles de nécrose apparaissent ci-dessous :
⊛ Nécrose coagulative : L’ischémie dans la plupart des organes, à l’exception du cerveau, peut entraîner une nécrose coagulative. Dans ce type de nécrose, l’architecture cellulaire reste préservée. Au microscope, les cellules apparaissent anucléées, éosinophiles, à structure préservée. Finalement, les cellules mortes sont éliminées par la phagocytose et les leucocytes.
⊛ Nécrose liquéfactive : Cette morphologie est le plus souvent observée dans le système nerveux central. Les cellules mourantes sont digérées par des enzymes hydrolytiques et perdent ainsi leur intégrité structurelle et se transforment en une masse visqueuse. Cette morphologie se produit également dans la plupart des infections bactériennes, et l’accumulation d’un tel matériel nécrotique est appelée pus.
⊛ Nécrose caséeuse : Le terme caséeux signifie « ressemblant à du fromage », ce qui fait référence à l’aspect blanchâtre de la zone nécrosée. Cette nécrose se produit dans l’infection tuberculeuse et la zone nécrotique est appelée granulome.
⊛ Nécrose gangreneuse : Il ne s’agit pas d’un schéma morphologique mais plutôt d’un terme clinique désignant la nécrose ischémique des membres. Il a deux types i) sec (ischémie conduisant à une nécrose coagulative) et ii) humide (ischémie avec infection bactérienne superposée conduisant à une nécrose liquéfactive).
⊛ Nécrose graisseuse : Ce type de nécrose survient dans la pancréatite aiguë. La libération d’enzymes pancréatiques conduit à la liquéfaction des cellules graisseuses dans la cavité péritonéale. Ces cellules graisseuses liquéfiées se combinent ensuite avec du calcium et sont identifiées comme des zones blanches crayeuses. Ce processus est appelé saponification. Au microscope, cela est visible sous forme de dépôts de calcium basophiles sur les contours des cellules graisseuses nécrotiques. Une nécrose graisseuse se produit également dans le tissu mammaire en raison de la saponification des graisses.
⊛ Nécrose fibrinoïde : Ce type de nécrose se produit dans les vaisseaux sanguins en raison du dépôt de complexes immuns dans les parois des vaisseaux sanguins, entraînant une fuite de fibrine. Cette coloration observée apparaît comme un matériau amorphe rose vif.
⊛ Autres : Le « red dead » neuron classique a été décrit comme la manifestation morphologique la plus courante de la mort neuronale dans une cellule nécrotique du système nerveux central. Mais il a été prouvé que la même cellule peut se produire à la suite d’une apoptose ou d’une nécrose. Puisqu’il existe des difficultés considérables pour différencier l’apoptose de la nécrose dans le rein et le cerveau, il est recommandé d’utiliser l’un ou l’autre terme s’il y a confiance (basée sur l’expérience ou des études spéciales) dans le type de mort cellulaire présent.
Il existe différents schémas de nécrose en fonction de la nature de l’atteinte et du tissu : Unicellulaire (cellules uniques non contiguës dans un tissu qui se caractérisent par un gonflement de la cellule et du noyau et un cytoplasme pâle), Focal (Contient des cellules contiguës), Multifocale, Diffuse (Contient des cellules contiguës), Centrilobulaire, et Zonal. Ainsi, la nécrose est une caractéristique de diverses maladies et les différents modèles de nécrose observés dans différents organes aident à identifier le mécanisme des lésions cellulaires et les éventuels stimuli nocifs qui les provoquent. Par exemple :
⊛ La nécrose tubulaire aiguë observée dans le rein en est un exemple. Divers médicaments ont été associés à des lésions rénales, notamment la phénylbutazone, l’ibuprofène et l’acide méfénamique.
⊛ De même, la consommation d’alcool a été étudiée pour entraîner une inflammation hépatique, une nécrose et une stéatose. L’inflammation a été proposée comme un événement de progression dans le développement de la stéatohépatite alcoolique.
⊛ L’ischémie cardiaque entraînant une lésion myocardique, l’ischémie cérébrale entraînant un accident vasculaire cérébral et l’ischémie des membres entraînant une gangrène sont tous des exemples cliniques de nécrose. La nécrose permet ainsi de décrire le mécanisme pathologique des maladies les plus couramment rencontrées.
Signification clinique :
L’identification des différents types de nécrose et de la cause sous-jacente de la nécrose peut aider à cibler le traitement de plusieurs maladies. La plupart du temps, identifier la cause de la nécrose et la traiter est plus important que d’enlever les tissus morts. Dans le cas de l’infarctus du myocarde, nous savons que la nécrose se produit en raison de l’hypoxie due à l’occlusion des vaisseaux coronaires. Par conséquent, le traitement vise à ouvrir les vaisseaux coronaires soit par thrombolyse, soit par PCI pour rétablir l’approvisionnement en sang. Aussi, une réponse inflammatoire accompagne la nécrose, il serait donc utile d’étudier les effets des anti-inflammatoires sur la suppression de la nécrose.
La nécrose comme mort cellulaire programmée
En règle générale, la nécrose n’est pas associée à l’activation de la caspase ou au développement normal, mais différents types de nécrose régulée ou programmée ont été décrits, tels que la nécroptose, la pyroptose et la ferroptose. La nécrose régulée synchronisée est le résultat d’une peroxydation lipidique spécifique dans la ferroptose. Comme l’apoptose, les voies enzymatiques telles que les caspases, les kinases et le système polyubiquitine jouent le rôle principal dans la nécroptose et la pyroptose.
Nécroptose ou nécrose apoptotique
La nécroptose est un mode alternatif de mort cellulaire régulée imitant les caractéristiques de l’apoptose et de la nécrose. La nécroptose est très fréquente in vivo, principalement dans diverses formes de neurodégénérescence et de mort infligées par ischémie ou infection. Contrairement à la nécrose non ordonnée, la nécroptose est un type de mort cellulaire plus physiologique et programmé et partage plusieurs processus clés avec l’apoptose et l’autophagie. La nécroptose est déclenchée par les membres de la famille du facteur de nécrose tumorale (TNF), les récepteurs de type Toll (TLR), l’interféron et certains autres médiateurs, et nécessite l’inhibition de la caspase 8 et l’assemblage du nécrosome (complexe RIPK1-RIPK3 IIb) – la protéine RIPK3 (reconnue comme régulateur de l’inflammation, de la survie cellulaire et de la maladie) et son substrat MLKL sont les acteurs cruciaux de cette voie. Après la liaison du TNF-α au TNFR1, il existe trois fonctions divergentes possibles, notamment la survie cellulaire, l’apoptose ou la nécroptose via différents complexes de signalisation. Le complexe I est pro-survie et le complexe IIa est pro-apoptotique.
Cela se produit en raison de l’activation du domaine kinase de la protéine 1 interagissant avec le récepteur (RIP1 ou Receptor-interacting serine/threonine-protein kinase 1 ou RIPK1) et de l’assemblage du complexe de signalisation contenant RIP1/RIP3. RIP3 régule la nécroptose de manière dépendante de RIP1 dans le diabète. Une fois recruté par RIP1 après exposition à HG, RIP3 est activé par auto-phosphorylation pour favoriser la phosphorylation du domaine kinase de lignée mixte comme la protéine (MLKL). Ensuite, MLKL oligomérise et se transloque vers la membrane cellulaire, interagit avec les lipides de phosphatidylinositol et la cadiolipine pour entraîner la perméabilisation de la membrane. La voie d’activation de la nécroptose après une stimulation élevée par le glucose est médiée par le ligand des récepteurs de la mort tels que le récepteur 1 du facteur de nécrose tumorale (TNFR1) et les récepteurs Fas. Les modèles moléculaires associés aux dommages (DAMP), la liaison aux nucléotides et les récepteurs de type oligomérisation (NOD) (NLR), le ripoptosome et les complexes de protéine kinase R (PKR) déclenchent également la nécroptose. La nécroptose médiée par le TNFR1 est la voie la plus étudiée pour activer la nécroptose.
Par ailleurs, la protéine kinase II dépendante de la calmoduline (CaMKII) est un substrat RIP3 nouvellement trouvé pour induire la nécroptose. CaMKII est abondant dans le myocarde et est inactivé dans des conditions normales. La phosphorylation de CaMKII par Ca2+ intracellulaire ou RIP1 facilite la nécroptose. Pendant ce temps, HG augmente également les ROS pour activer CaMKII par oxydation. Au total, les changements pathologiques ci-dessus déclenchent l’ouverture du pore de transition de perméabilité mitochondriale (mPTP), qui est impliqué dans la voie finale de la nécroptose.
La dérégulation de la nécroptose s’est avérée associée à des conditions pathologiques telles que le cancer, les maladies neurodégénératives et les maladies inflammatoires. Dans cet article chronologique, nous discutons des mécanismes moléculaires de la nécroptose et de sa pertinence pour les maladies.
Pyroptose
La pyroptose est une autre forme de nécrose programmée. Il est responsable de la lyse cellulaire et de la libération extracellulaire de cytokines pro-inflammatoires telles que l’interleukine-1β (IL-1β) et l’interleukine-18 (IL-18).
Il existe deux voies différentes, notamment la voie canonique et la voie non canonique dans la pyroptose. Dans la voie canonique, les complexes multiprotéiques cytoplasmiques, nommés inflammasome, sont constitués de la famille des récepteurs de type NOD (nucleotide-binding oligomerization domain) (NLR) (dont NLRP3, NLRP1, NLRC4, NLRP9 et NLRP6), des domaines pyrine et HIN (PYHIN ) familles de protéines (absentes dans le mélanome 2, AIM2) et protéines pyrines. Des inflammasomes distincts sont reconnus par des modèles moléculaires associés aux agents pathogènes (PAMP) ou motifs moléculaires associés aux dégâts (DAMP ou Damage Associated Molecular Pattern) pour activer la caspase 1, conduisant à la pyroptose.
Dans la voie non canonique, le lipopolysaccharide bactérien (LPS) est délivré au cytosol pour activer la caspase 11. La caspase 11 activée induit directement la pyroptose. La caspase 11 clivée active également la Gasdermin-D (GSDMD) pour former un pore membranaire. Pendant ce temps, l’inflammasome NLRP3 est activé par le fragment N-terminal de GSDMD (GSDMD-N) pour déclencher la pyroptose via la voie canonique.
Ferroptose
La ferroptose est une forme de mort cellulaire nécrotique qui a été décrite récemment grâce à l’utilisation d’un composé chimique, l’érastine, sélectionné sur sa capacité à tuer les cellules cancéreuses porteuses de l’oncogène Ras activé. Le concept de ferroptose met en lumière le rôle critique des adaptations du métabolisme des cellules cancéreuses, qui constituent peut-être un de leurs talons d’Achille.
Dans les cellules exposées aux inducteurs de ferroptose, des perturbations majeures du métabolisme redox engendrent des altérations de la perméabilité membranaire et une perte de viabilité cellulaire. Cette modalité unique de mort cellulaire, entraînée par la peroxydation des phospholipides de la membrane plasmique dépendant du fer, est régulée par de multiples voies métaboliques cellulaires, notamment l’homéostasie redox, la manipulation du fer, l’activité mitochondriale et le métabolisme des acides aminés, des lipides et des sucres, en plus de diverses voies de signalisation pertinentes pour la maladie. De nombreuses lésions d’organes et pathologies dégénératives sont provoquées par la ferroptose
Contrairement à l’apoptose, la ferroptose se déroule sans induction de la perméabilisation de la membrane externe des mitochondries. Il n’est donc pas observé de libération cytosolique du cytochrome c ou d’activation des caspases. En revanche, un stress oxydant intense apparaît précocement et joue un rôle essentiel dans la perte de la viabilité cellulaire. Des molécules anti-oxydantes à tropisme membranaire, comme le β-carotène ou l’α-tocophérol (vitamine E), protègent les cellules cancéreuses de la ferroptose induite par l’érastine. La déplétion des stocks intracellulaires en fer protège aussi efficacement les cellules de la mort induite par l’érastine. Le rôle du fer dans la ferroptose est conforme aux connaissances concernant la biologie de cet élément. Dans les systèmes biologiques, le fer libre permet la transformation des espèces oxydantes faiblement actives (telles que le peroxyde d’hydrogène, H2O2) en des oxydants beaucoup plus puissants, comme le radical hydroxyle (OH˙). Ce dernier est capable d’oxyder la plupart des constituants cellulaires, protéines, acides nucléiques et lipides insaturés (notamment les acides gras polyéthyléniques). Le pool intracellulaire de fer libre joue donc un rôle essentiel dans les mécanismes redox de la ferroptose.
Références
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol Rev. 2019 Oct 1;99(4):1765-1817. doi: 10.1152/physrev.00022.2018
Chen Y, Hua Y, Li X, Arslan IM, Zhang W and Meng G (2020) Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 11:42. doi: 10.3389/fphar.2020.00042
Dhuriya, Y.K., Sharma, D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation 15, 199 (2018). https://doi.org/10.1186/s12974-018-1235-0
Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005 Apr;73(4):1907-16. doi: 10.1128/IAI.73.4.1907-1916.2005.
Syntichaki P, Tavernarakis N. Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep. 2002 Jul;3(7):604-9. doi: 10.1093/embo-reports/kvf138.
Woo Y, Lee H-J, Jung YM, Jung Y-J. Regulated Necrotic Cell Death in Alternative Tumor Therapeutic Strategies. Cells. 2020; 9(12):2709. https://doi.org/10.3390/cells912270
Escobar, Ma., Echeverría, Olga, Vázquez-Nin, Gerardo. « Necrosis as Programmed Cell Death » In Cell Death: Autophagy, Apoptosis and Necrosis, edited by Tobias Ntuli. London: IntechOpen, 2015. doi : 10.5772/61483
Khalid N, Azimpouran M. Necrosis. [Updated 2022 Mar 9]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022.
et al. La ferroptose, une nouvelle forme de mort cellulaire applicable au traitement médical des cancers. Med Sci (Paris) 2014 ; 30 : 779–783.