{kind=link}
Le neuropaludisme cérébral, résulte d’une infection à Plasmodium falciparum et a un taux de mortalité élevé. Les survivants du neuropaludisme peuvent conserver des séquelles de neuropaludisme à vie, y compris des convulsions et des déficits neurocognitifs affectant profondément leur qualité de vie. Comme le parasite Plasmodium ne pénètre pas dans le cerveau, mais réside à l’intérieur des érythrocytes et est confiné à la lumière du système vasculaire cérébral, la neuropathogenèse conduisant à ces séquelles neurologiques n’est pas claire et sous-étudiée. Fait intéressant, la pathologie du neuropaludisme post-mortem diffère selon les régions du cerveau, comme l’apparition de points hémorragiques dans la matière blanche par rapport à la matière grise. Divers facteurs liés à l’hôte et au parasite contribuent au risque de neuropaludisme, notamment l’exposition à un jeune âge, la génétique liée au parasite et à l’hôte, la séquestration du parasite et l’étendue des réponses inflammatoires de l’hôte. Jusqu’à présent, plusieurs traitements d’appoint proposés n’ont pas été couronnés de succès dans le traitement du neuropaludisme, mais sont très nécessaires. La neuropathogenèse du neuropaludisme spécifique à la région entraînant des séquelles neurologiques est intrigante, mais pas suffisamment abordée dans la recherche. Une plus grande attention à cela pourrait conduire au développement de traitements d’appoint efficaces pour traiter les séquelles neurologiques du neuropaludisme.
Source : Schiess, N., Villabona-Rueda, A., Cottier, K.E. et al. Pathophysiology and neurologic sequelae of cerebral malaria. Malar J 19, 266 (2020). DOI: 10.1186/s12936-020-03336-z
Le paludisme est transmis par la piqûre de moustiques anophèles femelles infectés par Plasmodium. Elle reste l’une des maladies à transmission vectorielle les plus courantes, entraînant une morbidité et une mortalité élevées. Bien qu’il existe plusieurs espèces de Plasmodium susceptibles de provoquer des maladies, Plasmodium falciparum et Plasmodium vivax sont les deux principales espèces responsables de la plupart des complications chez l’homme, P. vivax étant plus répandu dans les pays d’Asie du Sud-Est et en Inde [1,2,3]. En 2018, il y avait environ 228 millions de cas de paludisme dans le monde, entraînant 405 000 décès [1]. Parmi ces décès, 67 % (272 000) concernaient des enfants de moins de 5 ans [1]. De multiples complications peuvent survenir à la suite d’une infection à P. falciparum, le neuropaludisme entraînant certains des taux de mortalité les plus élevés [1,4,5]. De plus, les patients qui survivent au neuropaludisme peuvent rester avec des séquelles post-neuropaludisme à vie, en particulier des déficits neurologiques, affectant la qualité de vie [6].
Le paludisme grave, dû à une infection à P. falciparum, se présente différemment chez les enfants que chez les adultes, notamment en ce qui concerne l’apparition de neuropaludisme. Alors que la mortalité pédiatrique du neuropaludisme serait inférieure à la mortalité adulte, le neuropaludisme pédiatrique est associée à un taux plus élevé de convulsions et de déficits neurocognitifs post-neuropaludisme [7, 8]. Ces écarts dans la présentation du neuropaludisme peuvent survenir en raison de différences dans le cerveau immature, y compris des différences dans les réponses de l’hôte du système vasculaire cérébral dans différentes régions du cerveau à la séquestration et à l’ampleur de l’inflammation. Cette revue se concentre sur les mécanismes immunopathophysiologiques sous-jacents du paludisme pédiatrique à P. falciparum et les séquelles neurologiques subséquentes, comme on le voit en Afrique subsaharienne.
Susceptibilité et résistance génétiques de l’hôte
Étant donné que plus d’un million d’enfants par an mouraient de P. falciparum rien qu’en Afrique avant le XXIe siècle [4], le paludisme est, d’un point de vue génétique, le moteur de l’évolution entraînant des maladies érythrocytaires génétiques telles que la drépanocytose. , thalassémie et déficit en glucose-6-phosphate déshydrogénase. Ceci est étayé par les observations selon lesquelles, malgré la mortalité homozygote, l’allèle HbS a une prévalence élevée dans les zones d’endémie palustre ainsi que par l’observation que des mutations génétiques indépendantes se sont développées dans différentes populations ethniques et géographiques [9]. D’autres facteurs génétiques de l’hôte contribuant à la susceptibilité au neuropaludisme comprennent des facteurs inflammatoires et des régions régulatrices, telles que les variants du récepteur de l’interféron de type 1 au Malawi [10], IL17 au Nigeria et IL4 et IL22 dans les populations du Mali [11,12]. En outre, des rapports antérieurs ont montré un rôle pour les variantes Kilifi d’adhésion intercellulaire moléculaire -1 (ICAM-1) dans neuropaludisme [13].
Une étude récente à Kilifi, au Kenya, a identifié 15 gènes associés à une augmentation du paludisme pédiatrique [14], et une étude indienne a identifié des polymorphismes du TNF [15]. En outre, des études épidémiologiques ont rapporté une association des résultats des infections paludéennes avec l’âge et l’exposition antérieure à des modifications épigénétiques [16,17,18,19]. Cela découle d’une découverte récente selon laquelle la production des métabolites du cycle de l’acide citrique, le succinate et le fumarate, augmentait pendant le paludisme grave, y compris le neuropaludisme. Ces métabolites peuvent servir de modulateurs d’enzymes épigénétiques, telles que les histones et les ADN déméthylases [20]. Il est de plus en plus évident que les infections parasitaires récurrentes, en invoquant l’hyperréactivité de la stimulation du ligand des récepteurs de type Toll (TLR), peuvent entraîner des modifications épigénétiques avec des phénotypes résistants au paludisme [21]. En effet, ces modifications épigénétiques ont été rapportées chez des enfants kenyans infectés par Plasmodium [16].
Les co-infections chez les patients pédiatriques neuropaludiques, comme le VIH, sont considérées comme des facteurs de risque indépendants de décès. Des études d’autopsie ont démontré une multiplication par deux des monocytes et des plaquettes intravasculaires chez les enfants infectés par le VIH décédés de neuropaludisme [22]. De plus, une présence accrue de lymphocytes T a été observée dans les cerveaux des patients neuropaludiques avec une co-infection par le VIH [23,24]. Il est probable que chez les patients co-infectés, la dérégulation immunitaire associée au VIH amplifie davantage les dommages pathologiques de neuropaludisme, entraînant une augmentation de l’afflux de lymphocytes T dans le cerveau [22,24,25]. Pris ensemble, divers facteurs de l’hôte contribuent à la sensibilité au paludisme grave et, bien qu’il existe des différences entre les régions, les facteurs associés à de fortes réponses immunitaires de l’hôte semblent essentiels.
Caractéristiques cliniques
Le neuropaludisme est la complication neurologique la plus sévère de l’infection à P. falciparum et est un syndrome clinique caractérisé par une altération de la conscience, le coma étant la manifestation la plus sévère [26]. Les caractéristiques cliniques du paludisme pédiatrique, y compris le neuropaludisme, impliquent une fièvre diurne récurrente, qui est produite après la libération du parasite lors de la rupture des globules rouges infectés par Plasmodium (PRBC), secondaire à la réplication asexuée et à la libération de cytokines [9,27]. Les patients atteints d’infection aiguë peuvent présenter une encéphalopathie neuropaludisme diffuse, un coma évolutif rapide et/ou des convulsions sans retour à la conscience. Dans certains cas, des signes neurologiques focaux sont présents [28]. Aux stades terminaux de la maladie, les enfants présentent souvent des signes de dysfonctionnement du tronc cérébral, tels que des réflexes pupillaires et cornéens anormaux, un regard dysconjugué et des schémas respiratoires irréguliers [28,29,30,31,32].
Bien que certaines séquelles, telles que la cécité corticale, s’améliorent avec le temps, les évaluations de suivi clinique à long terme chez les survivants pédiatriques de neuropaludisme ont montré une persistance élevée des séquelles neurologiques, y compris l’hémiplégie, l’ataxie, la parésie, les troubles convulsifs, les déficits du langage, les troubles du comportement, les troubles cérébraux graves. paralysie et troubles cognitifs [28,29,32,33]. Ces séquelles neurologiques peuvent entraîner une altération de la qualité de vie et une perte d’années de vie corrigées de l’incapacité. Les facteurs sous-jacents exacts qui jouent un rôle dans la neuropathogenèse conduisant à de mauvais résultats neurologiques chez les enfants ne sont pas clairs. Cependant, les résultats de l’autopsie ont établi que la séquestration intravasculaire des globules rouges infectés par Plasmodium est associée à des lésions périvasculaires, notamment des lésions axonales, une perte de myéline et une rupture de la barrière hémato-encéphalique (BHE) [34], comme le montre la Fig.1. Comment exactement la séquestration conduit à la rupture de BBB n’est pas claire.
Comme discuté plus tard, la séquestration associée aux facteurs solubles de Plasmodium peut avoir des effets à la fois directs et indirects sur l’intégrité de la BHE, qui peuvent être amplifiés par la tempête de cytokines et l’afflux de facteurs plasmatiques, y compris l’albumine, qui sont toxiques pour les neurones. Fait intéressant, chez les patients pédiatriques africains, la séquestration se produirait dans le système vasculaire cérébral quelle que soit la région, mais la pathologie post-mortem a révélé différentes réponses vasculaires de l’hôte [34,35]. Une prédominance de multiples lésions ponctuées hémorragiques est observée dans les zones de la substance blanche et du corps calleux, mais non visible dans d’autres régions du cerveau, telles que la matière grise ou les ganglions de la base [35]. De plus, l’adulte neuropaludique à P. falciparum présente également une prédominance de lésions ponctuées de la substance blanche, comme le montrent la pathologie post-mortem et l’imagerie par résonance magnétique (IRM) [36,37]. Pris ensemble, cela suggère que l’hétérogénéité phénotypique potentielle dans le système vasculaire local de l’hôte [38] qui peut attirer une séquestration différentielle des PRBC peut conduire à des réponses alternatives de l’hôte [39] (Figure 1).
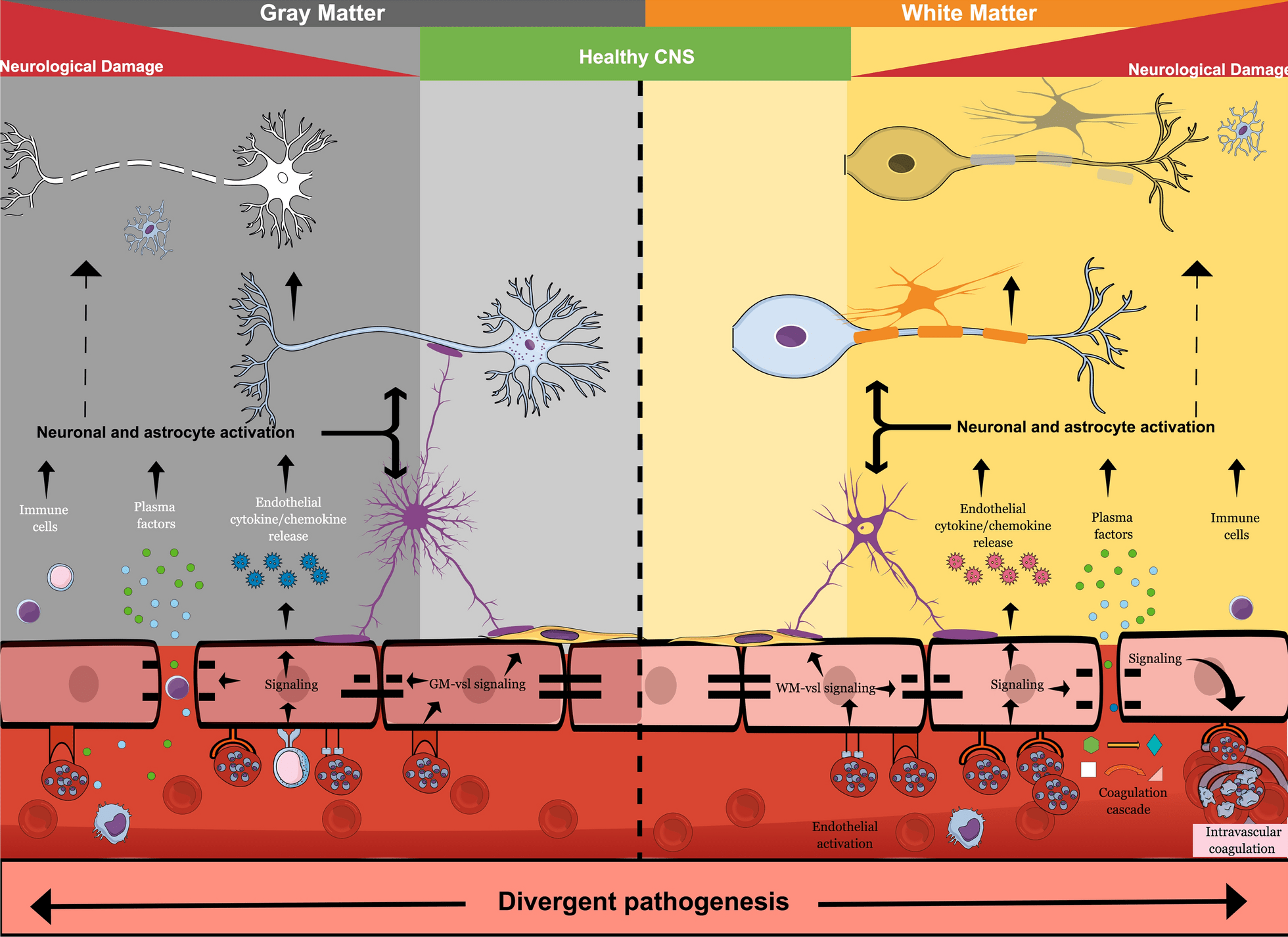
La pathologie du paludisme cérébral se manifeste différemment dans la substance blanche et la substance grise du cerveau. Alors que les points hémorragiques sont abondants dans la substance blanche, ils ne sont pas évidents dans la substance grise. La vascularisation cérébrale dans ces zones cérébrales est différente, ce qui peut conduire à une fixation différentielle de PRBC – guidée par l’expression du gène var – de PfEMP1 et à l’activation résultante de voies de signalisation alternatives dans la vascularisation endothéliale cérébrale de ces régions. La libération de chimiokines et de cytokines de l’endothélium BBB enflammé vers le cerveau, en conjonction avec l’ouverture de la barrière hémato-encéphalique qui permet l’entrée de substances plasmatiques neurotoxiques et de facteurs Plasmodium solubles dans le cerveau, conduit à l’activation astrogliale. Ceci, associé à un afflux de cellules immunitaires, provoque des dommages neurologiques responsables des séquelles neurologiques post-neuropaludisme.
Les critères de diagnostic cliniques de l’Organisation mondiale de la santé (OMS) pour le neuropaludisme (P. falciparum sur frottis sanguin, coma et aucune autre cause connue de coma) [40], peuvent entraîner des erreurs de diagnostic. En utilisant ces critères, une étude d’autopsie menée au Malawi [35] a rapporté que 23 % des cas de neuropaludisme diagnostiqués cliniquement étaient, en fait, une pathologie entièrement différente. Cela a le potentiel de fausser les résultats des études cognitives post-neuropaludisme dans la mesure où certains enfants pourraient déjà avoir des problèmes neurocognitifs préexistants. Il a été démontré que l’examen du fond d’œil et le diagnostic de rétinopathie améliorent la spécificité du diagnostic clinique de neuropaludisme, bien que la rétinopathie semble être moins spécifique chez l’adulte [8,41,42].
La rétinopathie du neuropaludisme est une constellation de changements oculaires qui comprend un blanchiment rétinien, des hémorragies rétiniennes, des changements vasculaires et un œdème papillaire, ainsi qu’une expression accrue de la molécule d’adhésion cellulaire vasculaire-1 (VCAM-1) [41, 43, 44]. La sévérité de la rétinopathie palustre est également positivement corrélée à un risque accru de décès [41, 43, 44]. Même après la résolution de l’infection à Plasmodium, les symptômes neurologiques persistent chez près d’un quart des enfants atteints de neuropaludisme avec rétinopathie [28]. De plus, il a été constaté que les patients neuropaludiques sans rétinopathie avaient des conditions neurologiques préexistantes, ce qui permet par la suite la possibilité d’évaluations neurologiques post-neuropaludisme inexactes [8]. Malheureusement, tous les cliniciens n’ont pas accès aux fonduscopes, car ces instruments sont relativement coûteux pour le LIMC. Accès accru à des adaptations de fond d’œil abordables, par ex. l’utilisation de téléphones cellulaires adaptés en combinaison avec des algorithmes et une formation appropriés améliorera le diagnostic approprié de neuropaludisme et peut fournir des prédictions sur le risque de séquelles neurologiques.
L’accès à d’autres modalités d’imagerie non invasives, telles que l’IRM, est extrêmement limité dans les pays à revenu faible ou intermédiaire. Néanmoins, les études IRM ont permis de mieux comprendre la pathogenèse et la cause de décès des patients pédiatriques neuropaludiques subsahariens. Les résultats de l’IRM chez 120 enfants atteints de neuropaludisme positif à la rétinopathie ont démontré une augmentation du volume cérébral (50%) et des anomalies cérébrales en T2, suggérant une inflammation des ganglions de la base (84,2%), de la substance blanche (71,7%), du tronc cérébral, du thalamus (40%), corps calleux (49,2%) et cervelet (49,2%) œdème vasogène, par opposition à cytotoxique. Des micro-hémorragies et une séquestration de parasites se sont produites dans les mêmes régions de substance blanche. Les résultats de l’imagerie pondérée en diffusion sont cohérents avec les micro-hémorragies et la séquestration parasitaire co-occurrentes dans les régions de la substance blanche avec congestion vasculaire [45]. Ces différences régionales dans les résultats de l’IRM suggèrent des différences potentielles dans la vascularisation de l’hôte entre ces régions de matière blanche et grise (Fig.1).
Les résultats de l’IRM qui ont été associés à des résultats de MC faibles à mortels comprennent des signes de pression intracrânienne élevée, un œdème cérébral, une diminution du volume de liquide céphalo-rachidien (LCR), une atteinte cérébrale postérieure, des lésions de la substance grise thalamique et supratentorielle et des zones inégales d’atteinte lobulaire [31,45]. Des études IRM ont également démontré un lien clair entre l’œdème cérébral, la profondeur du coma et l’augmentation de la mortalité [30].
Neuropathogenèse du paludisme cérébral
La pathogenèse du neuropaludisme est multiforme et, jusqu’à récemment, a été compliquée par l’hétérogénéité de la maladie, des classifications de cas cliniques souvent inexactes et un manque d’études cliniques prospectives à grande échelle [35]. Bien que déjà adoptée par certains groupes ayant accès à la copie du fond d’œil, une utilisation standard globale de la rétinopathie palustre comme critère d’inclusion pour les études entraînera probablement un suivi plus précis pour évaluer les séquelles tardives de le neuropaludisme. L’énigme de la neuropathogenèse du neuropaludisme et du coma qui en résulte a confondu les scientifiques pendant des décennies, car l’agent pathogène lui-même, résidant à l’intérieur des PRBC, ne pénètre pas directement ou physiquement dans le système nerveux central (SNC) en raison de la BHE, mais reste à l’intérieur de la lumière vasculaire (Fig.1). Pourtant, des symptômes neurologiques graves, y compris le coma, sont une caractéristique de le neuropaludisme et des preuves pathologiques de lésions neuronales ont également été démontrées par des niveaux élevés de tau dans le LCR des enfants atteints de neuropaludisme [46]. Cela met en évidence le rôle vital de l’endothélium BBB dans le neuropaludisme, car le BBB est à l’interface de la séquestration intravasculaire PRBC et des dommages neuronaux sous-jacents (Fig.1).
Deux théories principales (1) l’« hypothèse mécanique » et (2) l’hypothèse de la « tempête de cytokines » fournissent une explication sous-jacente à la neuropathogenèse du neuropaludisme. L’hypothèse mécanique est basée sur la contribution de la séquestration intravasculaire des PRBC qui entraîne de multiples conséquences, dont la congestion vasculaire, l’hypoperfusion et l’hypoxie localisée [47,48]. De plus, les différences de débit sanguin local peuvent contribuer à l’augmentation de la pression intracrânienne dans le neuropaludisme et aux différences lobulaires. Cela peut être dû soit à des différences d’apport vasculaire, par ex. lobe occipital via l’artère cérébrale postérieure par rapport aux autres lobes via le cercle de Willis, en raison de la séquestration des PRBC ou en raison d’une combinaison de ces facteurs. Ensemble, ces facteurs conduisent finalement à une rupture de la BHE, à un œdème cérébral et à un état pro-thrombotique [49,50,51]. La protéine-1 de la membrane érythrocytaire de P. falciparum codée par le parasite (PfEMP-1) est exprimée sur les surfaces PRBC et interagit avec les récepteurs de l’hôte.
PfEMP-1 est conçu pour sauver les PRBC de la clairance par la rate et est responsable de la séquestration intravasculaire des PRBC. PfEMP-1 est codé par un gène variable (gène var) et, selon le gène var exprimé, interagit avec divers récepteurs d’adhérence de l’hôte, tels que ICAM-1, EPCR et CD36 [52,53,54,55]. La liaison de PRBC exprimant le différentiel PfEMP-1, tel que codé par la famille de gènes var, à son récepteur respectif conduit à la signalisation de l’hôte en aval, y compris l’activation des voies inflammatoires et coagulantes, conduisant finalement à la perte de l’intégrité de la BBB et à l’encéphalopathie (Fig.1). De plus, comme évalué dans des expériences in vitro avec des cellules endothéliales cérébrales, la réactivité endothéliale différentielle de l’hôte peut affecter le développement du neuropaludisme chez les patients [56]. Les données post-mortem, les modèles animaux de neuropaludisme et les données in vitro démontrent que la séquestration des PRBC est corrélée à l’activation vasculaire cérébrale. Ceci est démontré par la présence de gros noyaux vésiculaires, la destruction endothéliale, l’activation du facteur de transcription NF-κB, l’expression accrue de molécules d’adhésion cellulaire, telles que ICAM-1, VCAM-1, la sélectine E, la libération de cytokines et la dégradation de BBB [35,57,58,59,60,61,62]. Les dommages endothéliaux dans le neuropaludisme sont également démontrés par des modifications du glycocalix endothélial lors de l’exposition au PRBC, à la fois in vitro [63] et in vivo dans le neuropaludisme humain [64] et le neuropaludisme expérimental murin [65] et la libération de vésicules endothéliales dans le circulation [66].
Bien que plusieurs études corrèlent le degré de séquestration des PRBC dans le cerveau à une sévérité accrue de le neuropaludisme [34,67,68], la mesure dans laquelle cela est corrélé aux symptômes cliniques, au développement du coma et à la mortalité dans le neuropaludisme est débattue [69]. Par conséquent, comme le propose «l’hypothèse de la tempête de cytokines (orage cytokinique)», l’inflammation périphérique, l’activation des neutrophiles [60] et l’augmentation de la circulation de plusieurs cytokines sériques telles que le TNF, l’IFNγ et l’IL-2, l’IL-6, l’IL-8 et l’IL-10 contribuent à la pathogenèse du neuropaludisme [12,70]. Par rapport aux patients atteints de paludisme non compliqué, l’IL-6 circulant, la MCP-1 et l’expression endothéliale vasculaire de CD61 sont régulées à la hausse [56]. L’augmentation des marqueurs inflammatoires indique à la fois une inflammation des cellules immunitaires et endothéliales et est associée à la séquestration des PRBC [57,61]. Des niveaux accrus de protéines plasmatiques solubles des neutrophiles et une chimiotaxie altérée des neutrophiles ont été trouvés dans les neuropaludismes pédiatriques avec rétinopathie [60], indiquant une activation des neutrophiles. Ces neutrophiles activés peuvent, comme les monocytes localisés intravasculaires [22], contribuer à l’activation vasculaire.
De plus, les cellules T CD8 ont été trouvées associées au système vasculaire cérébral, à la fois intravasculaire et périvasculaire et à la lame basale endothéliale où elles peuvent contribuer à l’activation vasculaire cérébrale, à la fois dans les études humaines et murines. Des études sur le neuropaludisme expérimental murin ont indiqué que les cellules T qui ont transmigré plus loin dans le neuropile peuvent endommager les neurones par la libération de Granzyme B et/ou de perforine [71,72]. À l’heure actuelle, il n’est pas clair si les cellules CD8+T envahissent préférentiellement des régions cérébrales spécifiques de la matière blanche ou de la matière grise. Des mécanismes supplémentaires par lesquels des dommages neuronaux se produisent peuvent impliquer des caspases dans certains neurones, comme le montre le neuropaludisme humain [73]. L’intégrité vasculaire et la transmigration lymphocytaire peuvent également être affectées par des altérations sphingolipidiques. Par exemple, les agents thérapeutiques bloquant la sphingosine-1-phosphate (S1P), tels que le FTY720, ont diminué le trafic lymphocytaire dans le cerveau et abaissé les niveaux périphériques d’IFNγ [74]. Toutes les études n’ont pas trouvé de relation entre les cytokines périphériques et l’œdème cérébral dans le neuropaludisme [75].
Pris ensemble, la séquestration et l’inflammation, associées à une élévation des facteurs de coagulation et à des altérations des métabolites sanguins, contribuent toutes à la neuropathogenèse du neuropaludisme [46,70,76,77,78,79,80,81], qui peut survenir dans une région- manière spécifique, par exemple matière blanche ou matière grise. Indépendamment de l’hypothèse de neuropaludisme médiée par la tempête de cytokines ou la séquestration, les effets de l’activation endothéliale peuvent être observés progressivement même dans les présentations subcliniques de la parasitémie, comme l’indiquent les taux sériques élevés de facteur von Willebrand (VWF), ICAM-1 soluble et non VCAM-1 soluble [82,83]. Cela signifie l’importance primordiale de la contribution de l’endothélium BBB dans la pathogenèse du neuropaludisme et bien que la relation exacte entre l’œdème cérébral et l’inflammation périphérique n’ait pas encore été complètement élucidée, il est probable qu’ils soient également corrélés avec les processus de réparation des cellules souches neurales en aval.
Neuro-séquelles post-neuropaludisme et mécanismes potentiels
Après le neuropaludisme, des séquelles neurologiques persistantes, notamment des troubles cognitifs, des capacités motrices, de la coordination visuelle, des convulsions et un trouble d’hyperactivité avec déficit de l’attention, surviennent chez jusqu’à 25 % des survivants pédiatriques [28,33,84,85,86]. Le risque le plus élevé de déficits du développement moteur, du langage et social concernait les enfants de moins de 5 ans [87]. Entre 3 et 6 mois après l’infection, les déficits cognitifs spécifiques à la mémoire de travail peuvent s’intensifier, le développement du langage étant le plus systématiquement affecté chez les survivants pédiatriques [7,32,88,89]. Les troubles cognitifs, y compris la mémoire et l’attention, peuvent persister jusqu’à neuf ans après les épisodes de neuropaludisme [84,85]. Dix pour cent des survivants pédiatriques de neuropaludisme dans une étude avaient au moins une séquelle de santé mentale avec un début allant de six à douze mois après l’infection et une médiane de 21 mois de suivi.
Les trois principaux troubles de santé mentale de ce groupe étaient le trouble déficitaire de l’attention avec hyperactivité, le trouble des conduites et le trouble oppositionnel avec provocation [90]. Après le neuropaludisme, des patients présentant des comportements extériorisés accrus (c’est-à-dire une mauvaise attention et de l’agressivité) ont également été signalés [87,91]. Cependant, l’évaluation précise des séquelles neurologiques peut être difficile. La plupart des études réalisées jusqu’à présent sur les séquelles à long terme de la MC ont utilisé la définition clinique de 2000 de l’OMS [7,84,85,88,91,92]. Cela pourrait signifier qu’une partie de ces enfants n’avaient pas de véritable neuropaludisme, ou une autre condition infectieuse sous-jacente également. Cela ouvre la possibilité que des enfants aient été inclus qui avaient déjà des conditions neurocognitives préexistantes ou que cela était dû à la co-infection et non au neuropaludisme. Quoi qu’il en soit, la présence de séquelles neurologiques chez les enfants positifs et négatifs à la rétinopathie a été bien documentée [29].
Les enfants sans rétinopathie présentent un schéma clinique et une présentation différents. Bien que les patients positifs pour la rétinopathie présentent plus d’anomalies à l’IRM dans une variété de régions cérébrales différentes, le pourcentage de patients présentant des séquelles neurologiques est similaire [30]. La mortalité plus faible chez les patients sans la rétinopathie peut être due à la présence de co-infections potentiellement protectrices, faussant les réponses immunitaires [30,45,93]. Il est intéressant de noter que diverses régions du cerveau semblent être différemment affectées. En raison de la diversité des cohortes d’études dans les différents pays et des méthodologies d’évaluation, ces études ne peuvent pas être directement comparées pour l’évaluation comportementale. Cependant, il est clair qu’à la suite d’infections compliquées à Plasmodium, il existe dans de nombreux cas un certain degré de difficultés comportementales qui satisfont aux critères de diagnostic d’un trouble de santé mentale.
Les convulsions sont fréquentes chez les enfants atteints de neuropaludisme et, comme conséquence à long terme, des troubles convulsifs soutenus, souvent réfractaires à au moins un médicament antiépileptique, peuvent se développer, même des mois après un épisode de neuropaludisme [28,29,33,84,85,94,95,96]. La présence de convulsions pourrait contribuer au développement d’autres séquelles neurologiques post-neuropaludisme, y compris des retards de développement [7,28]. En particulier, les crises aiguës pendant les épisodes de neuropaludisme étaient un indicateur de troubles du développement futurs soulevant la question de savoir si les crises aiguës pouvaient favoriser l’épileptogenèse et un risque accru d’épilepsie chronique [28,89,93].
L’élucidation des mécanismes sous-jacents au développement des troubles du comportement dans le neuropaludisme est un défi, d’autant plus que les comportements humains complexes sont difficiles à récapituler dans des modèles animaux. Il existe plusieurs modèles animaux qui sont utilisés pour étudier la pathogenèse de neuropaludisme, mais l’utilisation de modèles de primates pose des problèmes éthiques [97,98] et ceux-ci sont très coûteux. Le modèle animal le plus utilisé pour étudier le neuropaludisme est la souche ANKA de Plasmodium berghei dans un modèle murin, qui recrée un certain nombre de caractéristiques observées dans le neuropaludisme humain: notamment une inflammation vasculaire, des points hémorragiques dans certaines parties du cerveau et un ensemble de séquelles neurologiques qui peut être mesuré dans des tests comportementaux [99,100,101]. Comme tout système modèle pour une maladie, le modèle P. berghei ANKA n’est pas idéal et a reçu des critiques. Les critiques sur le neuropaludisme expérimental murin tournaient autour de l’absence d’un analogue de PfEMP-1 dans le parasite, d’une séquestration limitée, des découvertes de thérapeutiques efficaces dans le neuropaludisme expérimental murin par rapport à leur succès limité dans le neuropaludisme humain [102] et des différences perçues dans l’implication des lymphocytes T dans le neuropaludisme expérimental murin versus le neuropaludisme humain.
Bien que les auteurs mettent en garde à juste titre contre l’extrapolation aveugle des données de modèles murins, la prudence quant à leur position est justifiée, comme indiqué lors de plusieurs réunions scientifiques et dans des publications [103,104,105,106,107]. Il est important de noter que, bien que des travaux antérieurs n’aient pas réussi à montrer la présence de cellules CD8 + T dans les cerveaux neuropaludiques humains [34], récemment et grâce à des méthodologies mises à jour et très sensibles, leur présence a été confirmée dans les cerveaux neuropaludiques humains; ceux-ci étaient à la fois intravasculaires et périvasculaires, transmigrés dans le neuropile et associés au plexus choroïde, une porte d’entrée importante dans les ventricules et le liquide céphalo-rachidien [23,24,25,108]. En outre, les différences dans les résultats mesurés d’une étude peuvent également contribuer, par exemple, la survie ou l’incidence des séquelles neurologiques chez les survivants. Il est donc impératif de comparer les résultats obtenus dans des modèles animaux avec la pathologie humaine. En raison de problèmes éthiques et de directives réglementaires, la disponibilité d’échantillons humains pathologiques de neuropaludisme pour la recherche sur le neuropaludisme est très limitée. Une meilleure participation des patients/le consentement des membres de la famille et un meilleur accès à la communauté élargie de la recherche en neuropaludisme pourraient grandement profiter aux futurs patients. De plus, d’autres résultats que la simple survie doivent être pris en compte, tels que la réduction des séquelles neurologiques lors de l’évaluation de l’efficacité de nouveaux traitements adjuvants.
Plusieurs études utilisant le modèle murin à P. berghei ANKA ont démontré une augmentation des comportements anxieux après la résolution de l’infection à P. berghei ANKA [109]. Signalisation inflammatoire, telle que l’augmentation des niveaux de TNF cerveau-cortex en conjonction avec une augmentation de l’IL-6 ou de l’IL-1β, et des altérations des niveaux de facteur de croissance, par exemple le facteur neurotrophique dérivé du cerveau et la neuréguline peuvent contribuer au développement de l’anxiété dans le neuropaludisme expérimental murin et des séquelles comportementales [109,110,111,112]. Le peptide de la famille des endothélines ET-1 (pré-proendothéline) joue également un rôle important dans l’induction de dommages à la BHE et produit une image de neuropaludisme type dans les modèles de rongeurs [113]. Les antagonistes sélectifs des récepteurs de l’endothéline ont également amélioré les résultats du déclin cognitif et diminué les hémorragies cérébrales chez la souris [113,114,115]. En réponse à une inflammation accrue dans le neuropaludisme expérimental murin, Darling et al. ont montré la pertinence du récepteur tyrosine kinase EphA au niveau de l’endothélium de la BHE [116]. La régulation à la hausse d’EphA2 s’est avérée nécessaire pour la perte de protéines de jonction BBB à la fois dans le neuropaludisme expérimental murin et dans les cellules endothéliales microvasculaires du cerveau humain et pour l’infiltration de cellules CD8 + T dans le cerveau dans le neuropaludisme expérimental murin.
Prises ensemble, ces études suggèrent que les épisodes de neuropaludisme, tant chez l’homme que chez les modèles animaux, peuvent entraîner des déficits neurologiques. La présence de processus de réparation neuronale spécifiques après la résolution de l’infection à Plasmodium est impliquée, bien que la nature et l’efficacité de ces mécanismes sous-jacents ne soient pas claires. Les dommages neuronaux infligés lors d’une infection à Plasmodium pourraient être liés à l’étendue des réponses inflammatoires, à la séquestration des PRBC ou, après l’infection, à l’efficacité des mécanismes de réparation neurologique. On sait très peu de choses sur les processus de réparation post-neuropaludisme. Compte tenu de l’étendue des dommages neuronaux, les neuro-progéniteurs migreront et devront remplacer ces neurones endommagés. Notamment en raison de l’environnement encore très inflammatoire dans le cerveau, ce processus peut être biaisé et contribuer aux neuro-séquelles. Des recherches supplémentaires sur les mécanismes moléculaires pathogènes et de réparation sous-jacents de le neuropaludisme sont nécessaires. Cette recherche devrait se concentrer sur plusieurs domaines clés, notamment l’amélioration de l’inflammation vasculaire cérébrale, la rupture de la fonction barrière et les dommages neuronaux. En outre, il est nécessaire de se concentrer sur les processus de réparation neuronale dans le neuropaludisme. Cela peut conduire à l’identification de cibles pour des thérapies complémentaires ciblant le système vasculaire cérébral, la neuroprotection et la réparation neurologique. Les résultats de l’étude devraient se concentrer non seulement sur la survie, mais aussi cibler les séquelles neurologiques et le comportement.
À la recherche de nouvelles thérapies complémentaires
Des thérapies neuro-protectrices adjuvantes efficaces ne sont actuellement pas disponibles. De nombreuses tentatives pour développer des thérapies ciblant les séquelles neurologiques ont échoué ou ont même augmenté l’incidence des séquelles indésirables, comme l’ont revu Varo et al. [117]. Ces tentatives se sont principalement concentrées sur les efforts visant à réduire la séquestration, l’inflammation, la coagulation et le stress oxydatif et se sont jusqu’à présent avérées inefficaces pour réduire la mortalité et prévenir les effets neurologiques indésirables [118,119,120,121,122]. Par exemple, le traitement des patients avec de la dexaméthasone, couramment utilisée dans d’autres troubles neurologiques pour réduire l’inflammation et combattre l’œdème vasogénique, a en fait entraîné un coma plus long et une augmentation des complications neurologiques [123]. Les thérapies adjuvantes tentant de contrecarrer l’œdème cérébral avec le mannitol diurétique osmotique, connu pour abaisser la pression intracrânienne, n’ont montré aucun effet bénéfique chez les patients [124]. Ciblant également l’inflammation, le traitement par anticorps monoclonaux anti-TNF a aggravé les résultats de le neuropaludisme et augmenté l’incidence des séquelles neurologiques post-neuropaludisme [125].
Bien que la réduction pharmacologique des taux de TNF à l’aide de la pentoxifylline, un inhibiteur de la phosphodiestérase, ait montré de légères améliorations de la survie chez l’homme dans certaines études [126,127], d’autres n’ont montré aucun avantage sur les résultats cliniques [128,129], ou ont en fait augmenté la mortalité lorsqu’elles ont été étudiées chez les enfants [130]. Fait intéressant, des diminutions des niveaux de TNF et d’IL-6 dans l’hippocampe ont été observées dans un neuropaludisme expérimental murin avec un traitement au cannabidiol, ce qui a également augmenté les niveaux de facteur neurotrophique dérivé du cerveau, favorisé la survie de le neuropaludisme expérimental murin et empêché les comportements anxieux post-neuropaludisme [111]. Le double rôle du TNF dans la neurogenèse pourrait être dû à des effets différentiels de signalisation en aval, en fonction du récepteur du TNF impliqué. Alors que la signalisation via TNFR1 entrave de manière significative la neurogenèse, l’activation de TNFR2 contribue à la survie et à la prolifération des cellules souches neurales [131]. La neurogenèse, telle qu’elle est pilotée par les cellules souches neurales dans la zone sous-granulaire de l’hippocampe, est importante dans la fonction d’apprentissage et de mémoire [132,133].
Dans le neuropaludisme expérimental murin, une expression accrue de TNFR1 et, à un degré plus élevé, de TNFR2 est observée [134,135]. Par conséquent, un blocage complet de la signalisation du TNF peut entraver ses actions neurogènes et, par conséquent, conduire à des déficits neurocognitifs plus élevés. Le ciblage d’autres cytokines pourrait être prometteur pour d’éventuelles stratégies d’intervention. Une étude examinant le sérum post mortem et le LCR d’enfants au Ghana atteints de neuropaludisme a montré une élévation de IP-10, IL-8, MIP-1β, PDGFbb, IL-1ra, Fas-L, sTNF-R1 et sTNF-R2 [136]. La neuréguline, un facteur de croissance neurotrophique, s’est avérée protectrice de le neuropaludisme expérimental murin et a été postulée comme une thérapie d’appoint efficace pour réduire les lésions tissulaires du SNC [112]. L’administration de neuréguline-1 a entraîné une augmentation de 73 % de la survie dans le neuropaludisme expérimental murin, ainsi qu’une diminution des cytokines systémiques et du SNC ; TNF, IL-6, IL-1α et CXCL10 [137]. D’autres tentatives pour élucider le rôle d’autres contributeurs potentiels à l’amélioration de la survie comprennent le ciblage du système du complément avec un blocage des récepteurs C5/C5a [138] et la circulation sanguine/santé vasculaire à l’aide d’oxyde nitrique inhalé [139] L’oxyde nitrique joue un rôle dans la pathogenèse de neuropaludisme avec de faibles concentrations périphériques contribuant à la pathologie [140,141]. La thérapie d’appoint à base d’oxyde nitrique est également efficace dans le neuropaludisme expérimental murin par le biais de divers mécanismes, ce qui indique que l’administration directe d’oxyde nitrique ou par une supplémentation alimentaire en citrulline peut être bénéfique en tant que thérapie d’appoint [142].
D’autres thérapies complémentaires qui ciblent l’inflammation et les réponses immunitaires et qui sont prometteuses dans les études de neuropaludisme expérimental murin incluent la rosiglitazone, agoniste du récepteur gamma activé par les proliférateurs de peroxysomes, qui améliore à la fois la survie et les résultats neurocognitifs [143, 144]. L’atorvastatine, un médicament hypocholestérolémiant [145], atténue également l’inflammation et réduit ainsi à la fois les dommages endothéliaux cérébraux et l’ouverture de la BHE [146]. De plus, la rosglitazone a stimulé les voies neuroprotectrices et, dans les études sur l’homme, s’est révélée prometteuse pour le traitement du paludisme non compliqué [117], mais ses effets sur le neuropaludisme humain ne sont pas encore clairs. L’érythropoïétine, une hormone produite par les reins, a été envisagée pour une utilisation dans le neuropaludisme, ainsi que des composants antioxydants et anti-inflammatoires dans des modèles animaux. Des études murines ont démontré des résultats neuroprotecteurs positifs, soit seuls [147], soit associés à l’artésunate [148]. Cependant, des études cliniques ont démontré que des taux élevés d’érythropoïétine sont associés à un coma prolongé et à une mortalité accrue [149]. Comme l’inflammation de la BHE et la perte d’intégrité jouent également un rôle central dans le neuropaludisme, une approche thérapeutique potentielle consistant à bloquer l’EphA2 pour protéger la BHE de la dégradation a été suggérée dans des études récentes sur de neuropaludisme expérimental murin [116].
Il ressort clairement des études animales et cliniques que, bien que des niveaux élevés d’inflammation et d’œdème cérébral aient été associés à la mortalité et aux séquelles neurologiques de neuropaludisme, la résolution de ce problème sur le plan pharmacologique présente toujours des défis importants. La suppression complète de l’inflammation est préjudiciable, mais l’amélioration de l’inflammation semble bénéfique pour le résultat de survie de neuropaludisme et peut également être neuroprotectrice. Bon nombre de ces études sur les thérapies complémentaires chez la souris ont examiné les résultats de mortalité et n’ont pas évalué les troubles neurocognitifs. Ainsi, il est possible que des effets neurocognitifs aient été manqués. Ces types d’études auraient pu renforcer l’argument en faveur d’un traitement d’appoint. Des recherches supplémentaires sont nécessaires pour élucider la neuropathologie complexe conduisant à des déficits neurologiques à long terme, pour identifier des biomarqueurs prédictifs de la gravité des neuro-séquelles et comment cela peut être traité cliniquement avec des traitements neuroprotecteurs adjuvants. Cela nécessite de tester des résultats supplémentaires au-delà de la survie, à court et à long terme, et de mettre l’accent sur la réduction des séquelles neurologiques.
Remarques finales
Le neuropaludisme est une maladie dévastatrice avec une neuro-pathophysiologie complexe qui peut entraîner diverses séquelles neurologiques affectant une personne tout au long de sa vie. L’hétérogénéité des symptômes cliniques et des résultats va du rétablissement complet à diverses séquelles neurologiques et, souvent, au décès. Avec l’amélioration des soins de santé en Afrique subsaharienne, davantage de patients pédiatriques devraient survivre. Cette population peut développer des séquelles neurologiques durables, contribuant ainsi à un problème de santé mondial croissant. Plusieurs études cliniques ont identifié des caractéristiques spécifiques de ces séquelles neurologiques, notamment des déficits cognitifs et comportementaux et des troubles convulsifs. Les études post-mortem et d’imagerie humaines ont permis de mieux comprendre la neuropathologie. Les études de neuropaludisme expérimental murines peuvent aborder les mécanismes impliqués dans le développement de le neuropaludisme expérimental murin et la genèse des séquelles neurologiques. Bien que les recherches actuelles suggèrent un rôle important de l’inflammation dans l’apparition de lésions neuronales et le développement de séquelles post-neuropaludisme, des recherches supplémentaires sont nécessaires pour aborder les moteurs physiopathologiques moléculaires sous-jacents plus spécifiques et les mécanismes de signalisation. Cela souligne l’importance de la collaboration entre différents domaines, y compris la recherche clinique, la recherche animale et l’échange d’échantillons comme clé pour faire progresser les connaissances existantes sur la neuropathogenèse du neuropaludisme. Ensemble, cela peut conduire à l’identification de nouvelles cibles pour des traitements d’appoint afin d’améliorer les séquelles neurologiques post-neuropaludisme.
Références :
- WHO. Malaria Key Facts. Geneva, World Health Organization, 2019. https://www.who.int/news-room/fact-sheets/detail/malaria. Accessed 08 May 2019.
- Kochar DK, Das A, Kochar SK, Saxena V, Sirohi P, Garg S, et al. Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am J Trop Med Hyg. 2009;80:194–8. PubMed Google Scholar
- Battle KE, Lucas TCD, Nguyen M, Howes RE, Nandi AK, Twohig KA, et al. Mapping the global endemicity and clinical burden of Plasmodium vivax, 2000-17: a spatial and temporal modelling study. Lancet. 2019;394:332–43. PubMed PubMed Central Google Scholar
- Gething PW, Casey DC, Weiss DJ, Bisanzio D, Bhatt S, Cameron E, et al. Mapping Plasmodium falciparum mortality in Africa between 1990 and 2015. N Engl J Med. 2016;375:2435–45.
- Weiss DJ, Lucas TCD, Nguyen M, Nandi AK, Bisanzio D, Battle KE, et al. Mapping the global prevalence, incidence, and mortality of Plasmodium falciparum, 2000–17: a spatial and temporal modelling study. Lancet. 2019;394:322–31. PubMed PubMed Central Google Scholar
- Severe malaria. Trop Med Int Health. 2014;19 Suppl 1:7-131.
- Boivin MJ, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, et al. Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics. 2007;119:e360–6. PubMed PubMed Central Google Scholar
- Birbeck GL, Beare N, Lewallen S, Glover SJ, Molyneux ME, Kaplan PW, et al. Identification of malaria retinopathy improves the specificity of the clinical diagnosis of cerebral malaria: findings from a prospective cohort study. Am J Trop Med Hyg. 2010;82:231–4. PubMed PubMed Central Google Scholar
- Phillips MA, Burrows JN, Manyando C, van Huijsduijnen RH, Van Voorhis WC, Wells TNC. Malaria. Nat Rev Dis Primers. 2017;3:17050. PubMed Google Scholar
- Feintuch CM, Tare A, Cusumano LR, Benayoun J, Ryu S, Sixpence A, et al. Type I interferon receptor variants in gene regulatory regions are associated with susceptibility to cerebral malaria in Malawi. Am J Trop Med Hyg. 2018;98:1692–8. CAS PubMed PubMed Central Google Scholar
- Marquet S, Conte I, Poudiougou B, Argiro L, Dessein H, Couturier C, et al. A functional IL22 polymorphism (rs2227473) is associated with predisposition to childhood cerebral malaria. Sci Rep. 2017;7:41636. CAS PubMed PubMed Central Google Scholar
- Hunt NH, Ball HJ, Hansen AM, Khaw LT, Guo J, Bakmiwewa S, et al. Cerebral malaria: gamma-interferon redux. Front Cell Infect Microbiol. 2014;4:113. PubMed PubMed Central Google Scholar
- Jenkins NE, Mwangi TW, Kortok M, Marsh K, Craig AG, Williams TN. A polymorphism of intercellular adhesion molecule-1 is associated with a reduced incidence of nonmalarial febrile illness in Kenyan children. Clin Infect Dis. 2005;41:1817–9. CAS PubMed PubMed Central Google Scholar
- Ndila CM, Uyoga S, Macharia AW, Nyutu G, Peshu N, Ojal J, et al. Human candidate gene polymorphisms and risk of severe malaria in children in Kilifi, Kenya: a case-control association study. Lancet Haematol. 2018;5:e333–45. PubMed PubMed Central Google Scholar
- Mohanty S, Singh US, Mohanty S, Mohanty AK, Pande V, Das A. Evolutionary interplay of single nucleotide polymorphisms at the promoter region of TNF-alpha gene in different clinical outcomes of malaria in India. Infect Genet Evol. 2019;69:107–16. CAS PubMed Google Scholar
- Schrum JE, Crabtree JN, Dobbs KR, Kiritsy MC, Reed GW, Gazzinelli RT, et al. Cutting edge: plasmodium falciparum induces trained innate immunity. J Immunol. 2018;200:1243–8. CAS PubMed PubMed Central Google Scholar
- Qidwai T, Jamal F, Singh S. Exploring putative molecular mechanisms of human pyruvate kinase enzyme deficiency and its role in resistance against Plasmodium falciparum malaria. Interdiscip Sci. 2014;6:158–66. CAS PubMed Google Scholar
- Quin JE, Bujila I, Cherif M, Sanou GS, Qu Y, Vafa Homann M, et al. Major transcriptional changes observed in the fulani, an ethnic group less susceptible to malaria. Elife. 2017;6:e29156. PubMed PubMed Central Google Scholar
- Gupta H, Chaudhari S, Rai A, Bhat S, Sahu PK, Hande MH, et al. Genetic and epigenetic changes in host ABCB1 influences malaria susceptibility to Plasmodium falciparum. PLoS ONE. 2017;12:e0175702.PubMed PubMed Central Google Scholar
- Arts RJ, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab. 2016;24:807–19. CAS PubMed PubMed Central Google Scholar
- Franklin BS, Parroche P, Ataide MA, Lauw F, Ropert C, de Oliveira RB, et al. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci USA. 2009;106:5789–94. CAS PubMed Google Scholar
- Hochman SE, Madaline TF, Wassmer SC, Mbale E, Choi N, Seydel KB, et al. Fatal pediatric cerebral malaria is associated with intravascular monocytes and platelets that are increased with HIV coinfection. MBio. 2015;6:e01390. CAS PubMed PubMed Central Google Scholar
- Swanson PA, Hart GT, Russo MV, Nayak D, Yazew T, Pena M, et al. CD8+T cells induce fatal brainstem pathology during cerebral malaria via luminal antigen-specific engagement of brain vasculature. PLoS Pathog. 2016;12:e1006022. PubMed PubMed Central Google Scholar
- Riggle BA, Manglani M, Maric D, Johnson KR, Lee MH, Neto OLA, et al. CD8+T cells target cerebrovasculature in children with cerebral malaria. J Clin Invest. 2020;130:1128–38. CAS PubMed PubMed Central Google Scholar
- Barrera V, Haley MJ, Strangward P, Attree E, Kamiza S, Seydel KB, et al. Comparison of CD8(+) T cell accumulation in the brain during human and murine cerebral malaria. Front Immunol. 2019;10:1747. CAS PubMed PubMed Central Google Scholar
- Idro R, Marsh K, John CC, Newton CR. Cerebral malaria: mechanisms of brain injury and strategies for improved neurocognitive outcome. Pediatr Res. 2010;68:267–74. PubMed PubMed Central Google Scholar
- Karunaweera ND, Grau GE, Gamage P, Carter R, Mendis KN. Dynamics of fever and serum levels of tumor necrosis factor are closely associated during clinical paroxysms in Plasmodium vivax malaria. Proc Natl Acad Sci USA. 1992;89:3200–3. CAS PubMed Google Scholar
- Birbeck GL, Molyneux ME, Kaplan PW, Seydel KB, Chimalizeni YF, Kawaza K, et al. Blantyre malaria project epilepsy study (BMPES) of neurological outcomes in retinopathy-positive paediatric cerebral malaria survivors: a prospective cohort study. Lancet Neurol. 2010;9:1173–81. PubMed PubMed Central Google Scholar
- Postels DG, Taylor TE, Molyneux M, Mannor K, Kaplan PW, Seydel KB, et al. Neurologic outcomes in retinopathy-negative cerebral malaria survivors. Neurology. 2012;79:1268–72. PubMed PubMed Central Google Scholar
- Potchen MJ, Kampondeni SD, Seydel KB, Birbeck GL, Hammond CA, Bradley WG, et al. Acute brain MRI findings in 120 Malawian children with cerebral malaria: new insights into an ancient disease. Am J Neuroradiol. 2012;33:1740–6. CAS PubMed Google Scholar
- Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, et al. Brain swelling and death in children with cerebral malaria. N Engl J Med. 2015;372:1126–37. CAS PubMed PubMed Central Google Scholar
- Idro R, Carter JA, Fegan G, Neville BG, Newton CR. Risk factors for persisting neurological and cognitive impairments following cerebral malaria. Arch Dis Child. 2006;91:142–8. CAS PubMed Google Scholar
- Kariuki SM, Abubakar A, Newton CR, Kihara M. Impairment of executive function in Kenyan children exposed to severe falciparum malaria with neurological involvement. Malar J. 2014;13:365. PubMed PubMed Central Google Scholar
- Dorovini-Zis K, Schmidt K, Huynh H, Fu W, Whitten RO, Milner D, et al. The neuropathology of fatal cerebral malaria in Malawian children. Am J Pathol. 2011;178:2146–58. PubMed PubMed Central Google Scholar
- Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, Fosiko NG, et al. Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med. 2004;10:143–5. CAS PubMed Google Scholar
- Nagatake T, Hoang VT, Tegoshi T, Rabbege J, Ann TK, Aikawa M. Pathology of falciparum malaria in Vietnam. Am J Trop Med Hyg. 1992;47:259–64. CAS PubMed Google Scholar
- Wijdicks EFM, Park JG. Surviving cerebral malaria. Neurology. 2018;91:978–9. PubMed Google Scholar
- Villabona-Rueda A, Erice C, Pardo CA, Stins MF. The evolving concept of the blood brain barrier (BBB): from a single static barrier to a heterogeneous and dynamic relay center. Front Cell Neurosci. 2019;13:405. CAS PubMed PubMed Central Google Scholar
- Claessens A, Rowe JA. Selection of plasmodium falciparum parasites for cytoadhesion to human brain endothelial cells. J Vis Exp. 2012;59:e3122. Google Scholar
- World Health Organization. Communicable Diseases Cluster Severe falciparum malaria. Trans R Soc Trop Med Hyg. 2000;94(Suppl 1):S1–90. Google Scholar
- Barrera V, Hiscott PS, Craig AG, White VA, Milner DA, Beare NA, et al. Severity of retinopathy parallels the degree of parasite sequestration in the eyes and brains of Malawian children with fatal cerebral malaria. J Infect Dis. 2015;211:1977–86. PubMed Google Scholar
- Kochar DK, Shubhakaran A, Kumawat BL, Thanvi I, Joshi A, Vyas SP. Ophthalmoscopic abnormalities in adults with falciparum malaria. QJM. 1998;91:845–52. CAS PubMed Google Scholar
- Beare NA, Southern C, Chalira C, Taylor TE, Molyneux ME, Harding SP. Prognostic significance and course of retinopathy in children with severe malaria. Arch Ophthalmol. 2004;122:1141–7. PubMed Google Scholar
- MacCormick IJ, Beare NA, Taylor TE, Barrera V, White VA, Hiscott P, et al. Cerebral malaria in children: using the retina to study the brain. Brain. 2014;137:2119–42. PubMed PubMed Central Google Scholar
- Potchen MJ, Kampondeni SD, Seydel KB, Haacke EM, Sinyangwe SS, Mwenechanya M, et al. 1.5 tesla magnetic resonance imaging to investigate potential etiologies of brain swelling in pediatric cerebral malaria. Am J Trop Med Hyg. 2018;98:497–504. CAS PubMed PubMed Central Google Scholar
- Medana IM, Idro R, Newton CR. Axonal and astrocyte injury markers in the cerebrospinal fluid of Kenyan children with severe malaria. J Neurol Sci. 2007;258:93–8. PubMed Google Scholar
- White NJ, Warrell DA, Looareesuwan S, Chanthavanich P, Phillips RE, Pongpaew P. Pathophysiological and prognostic significance of cerebrospinal-fluid lactate in cerebral malaria. Lancet. 1985;1:776–8. CAS PubMed Google Scholar
- Macherson GG, Warrell MJ, White NJ, Looareesuwan S, Warrell DA. Human cerebral malaria A quantitative ultrastructural analysis of parasitized erythrocyte sequestration. Am J Pathol. 1985;119:385–401. Google Scholar
- Hemmer CJ, Kern P, Holst FG, Radtke KP, Egbring R, Bierhaus A, et al. Activation of the host response in human Plasmodium falciparum malaria: relation of parasitemia to tumor necrosis factor/cachectin, thrombin-antithrombin III, and protein C levels. Am J Med. 1991;91:37–44.CAS PubMed Google Scholar
- Jimmy EO, Saliu I, Ademowo O. Fibrinopeptide-A and fibrinogen interactions in acute, Plasmodium falciparum malaria. Ann Trop Med Parasitol. 2003;97:879–81. CAS PubMed Google Scholar
- Clemens R, Pramoolsinsap C, Lorenz R, Pukrittayakamee S, Bock HL, White NJ. Activation of the coagulation cascade in severe falciparum malaria through the intrinsic pathway. Br J Haematol. 1994;87:100–5. CAS PubMed Google Scholar
- Avril M, Bernabeu M, Benjamin M, Brazier AJ, Smith JD. Interaction between endothelial protein C receptor and intercellular adhesion molecule 1 to mediate binding of Plasmodium falciparum-infected erythrocytes to endothelial cells. MBio. 2016;7:e00615–6. CAS PubMed PubMed Central Google Scholar
- Brown A, Turner L, Christoffersen S, Andrews KA, Szestak T, Zhao Y, et al. Molecular architecture of a complex between an adhesion protein from the malaria parasite and intracellular adhesion molecule 1. J Biol Chem. 2013;288:5992–6003. CAS PubMed PubMed Central Google Scholar
- Lau CK, Turner L, Jespersen JS, Lowe ED, Petersen B, Wang CW, et al. Structural conservation despite huge sequence diversity allows EPCR binding by the PfEMP1 family implicated in severe childhood malaria. Cell Host Microbe. 2015;17:118–29. CAS PubMed PubMed Central Google Scholar
- Shabani E, Hanisch B, Opoka RO, Lavstsen T, John CC. Plasmodium falciparum EPCR-binding PfEMP1 expression increases with malaria disease severity and is elevated in retinopathy negative cerebral malaria. BMC Med. 2017;15:183. PubMed PubMed Central Google Scholar
- Wassmer SC, Moxon CA, Taylor T, Grau GE, Molyneux ME, Craig AG. Vascular endothelial cells cultured from patients with cerebral or uncomplicated malaria exhibit differential reactivity to TNF. Cell Microbiol. 2011;13:198–209. CAS PubMed PubMed Central Google Scholar
- Armah H, Dodoo AK, Wiredu EK, Stiles JK, Adjei AA, Gyasi RK, et al. High-level cerebellar expression of cytokines and adhesion molecules in fatal, paediatric, cerebral malaria. Ann Trop Med Parasitol. 2005;99:629–47. CAS PubMed Google Scholar
- Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes decrease the integrity of human blood-brain barrier endothelial cell monolayers. J Infect Dis. 2007;195:942–50. PubMed Google Scholar
- Tripathi AK, Sullivan DJ, Stins MF. Plasmodium falciparum-infected erythrocytes increase intercellular adhesion molecule 1 expression on brain endothelium through NF-kappaB. Infect Immun. 2006;74:3262–70. CAS PubMed PubMed Central Google Scholar
- Feintuch CM, Saidi A, Seydel K, Chen G, Goldman-Yassen A, Mita-Mendoza NK, et al. Activated neutrophils are associated with pediatric cerebral malaria vasculopathy in malawian children. MBio. 2016;7:e01300–15. CAS PubMed PubMed Central Google Scholar
- Brown H, Rogerson S, Taylor T, Tembo M, Mwenechanya J, Molyneux M, et al. Blood-brain barrier function in cerebral malaria in Malawian children. Am J Trop Med Hyg. 2001;64:207–13. CAS PubMed Google Scholar
- Turner G. Cerebral malaria. Brain Pathol. 1997;7:569–82. CAS PubMed Google Scholar
- Introini V, Carciati A, Tomaiuolo G, Cicuta P, Guido S. Endothelial glycocalyx regulates cytoadherence in Plasmodium falciparum malaria. J R Soc Interface. 2018;15:20180773. PubMed PubMed Central Google Scholar
- Yeo TW, Bush PA, Chen Y, et al. Glycocalyx breakdown is increased in African children with cerebral and uncomplicated falciparum malaria. FASEB J. 2019;33:14185–93. CAS PubMed Google Scholar
- Hempel C, Sporring J, Kurtzhals JAL. Experimental cerebral malaria is associated with profound loss of both glycan and protein components of the endothelial glycocalyx. FASEB J. 2019;33:2058–71. CAS PubMed Google Scholar
- Sierro F, Grau GER. The ins and outs of cerebral malaria pathogenesis: immunopathology, extracellular vesicles, immunometabolism, and trained immunity. Front Immunol. 2019;10:830. CAS PubMed PubMed Central Google Scholar
- Pongponratn E, Riganti M, Punpoowong B, Aikawa M. Microvascular sequestration of parasitized erythrocytes in human falciparum malaria: a pathological study. Am J Trop Med Hyg. 1991;44:168–75. CAS PubMed Google Scholar
- Ponsford MJ, Medana IM, Prapansilp P, et al. Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J Infect Dis. 2012;205:663–71. PubMed Google Scholar
- Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT, et al. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am J Pathol. 1999;155:395–410. CAS PubMed PubMed Central Google Scholar
- Mandala WL, Msefula CL, Gondwe EN, Drayson MT, Molyneux ME, MacLennan CA. Cytokine profiles in Malawian children presenting with uncomplicated malaria, severe malarial anemia, and cerebral malaria. Clin Vaccine Immunol. 2017;24:e00533–616. CAS PubMed PubMed Central Google Scholar
- Eeka P, Phanithi PB. Cytotoxic T lymphocyte granzyme-b mediates neuronal cell death during Plasmodium berghei ANKA induced experimental cerebral malaria. Neurosci Lett. 2018;664:58–65. CAS PubMed Google Scholar
- Huggins MA, Johnson HL, Jin F, et al. Perforin expression by CD8 T cells is sufficient to cause fatal brain edema during experimental cerebral malaria. Infect Immun. 2017;85:e00985–1016. CAS PubMed PubMed Central Google Scholar
- Medana IM, Mai NT, Day NP, Hien TT, Bethell D, Phu NH, et al. Cellular stress and injury responses in the brains of adult Vietnamese patients with fatal Plasmodium falciparum malaria. Neuropathol Appl Neurobiol. 2001;27:421–33. CAS PubMed Google Scholar
- Finney CA, Hawkes CA, Kain DC, Dhabangi A, Musoke C, Cserti-Gazdewich C, et al. S1P is associated with protection in human and experimental cerebral malaria. Mol Med. 2011;17:717–25. CAS PubMed PubMed Central Google Scholar
- Harawa V, Njie M, Kessler A, Choko A, Kumwenda B, Kampondeni S, et al. Brain swelling is independent of peripheral plasma cytokine levels in Malawian children with cerebral malaria. Malar J. 2018;17:435. CAS PubMed PubMed Central Google Scholar
- Gupta S, Seydel K, Miranda-Roman MA, Feintuch CM, Saidi A, Kim RS, et al. Extensive alterations of blood metabolites in pediatric cerebral malaria. PLoS ONE. 2017;12:e0175686. PubMed PubMed Central Google Scholar
- McDonald CR, Conroy AL, Hawkes M, Elphinstone RE, Gamble JL, Hayford K, et al. Brain-derived neurotrophic factor is associated with disease severity and clinical outcome in Ugandan children admitted to hospital with severe malaria. Pediatr Infect Dis J. 2017;36:146–50. PubMed Google Scholar
- Molyneux ME, Engelmann H, Taylor TE, Wirima JJ, Aderka D, Wallach D, et al. Circulating plasma receptors for tumour necrosis factor in Malawian children with severe falciparum malaria. Cytokine. 1993;5:604–9. CAS PubMed Google Scholar
- Moussa EM, Huang H, Thezenas ML, Fischer R, Ramaprasad A, Sisay-Joof F, et al. Proteomic profiling of the plasma of Gambian children with cerebral malaria. Malar J. 2018;17:337. PubMed PubMed Central Google Scholar
- Punsawad C, Viriyavejakul P. Reduction in serum sphingosine 1-phosphate concentration in malaria. PLoS ONE. 2017;12:e0180631. PubMed PubMed Central Google Scholar
- Esamai F, Ernerudh J, Janols H, Welin S, Ekerfelt C, Mining S, et al. Cerebral malaria in children: serum and cerebrospinal fluid TNF-alpha and TGF-beta levels and their relationship to clinical outcome. J Trop Pediatr. 2003;49:216–23. PubMed Google Scholar
- Park GS, Ireland KF, Opoka RO, John CC. Evidence of endothelial activation in asymptomatic Plasmodium falciparum parasitemia and effect of blood group on levels of von Willebrand factor in malaria. J Pediatric Infect Dis Soc. 2012;1:16–25. PubMed PubMed Central Google Scholar
- Phiri HT, Bridges DJ, Glover SJ, van Mourik JA, de Laat B, M’baya B, et al. Elevated plasma von Willebrand factor and propeptide levels in Malawian children with malaria. PLoS ONE. 2011;6:e25626. CAS PubMed PubMed Central Google Scholar
- Carter JA, Ross AJ, Neville BG, Obiero E, Katana K, Mung’ala-Odera V, et al. Developmental impairments following severe falciparum malaria in children. Trop Med Int Health. 2005;10:3–10. PubMed Google Scholar
- Carter JA, Mung’ala-Odera V, Neville BG, Murira G, Mturi N, Musumba C, et al. Persistent neurocognitive impairments associated with severe falciparum malaria in Kenyan children. J Neurol Neurosurg Psychiatry. 2005;76:476–81. CAS PubMed PubMed Central Google Scholar
- Oluwayemi IO, Brown BJ, Oyedeji OA, Oluwayemi MA. Neurological sequelae in survivors of cerebral malaria. Pan Afr Med J. 2013;15:88. PubMed PubMed Central Google Scholar
- Brim R, Mboma S, Semrud-Clikeman M, Kampondeni S, Magen J, Taylor T, et al. Cognitive outcomes and psychiatric symptoms of retinopathy-positive cerebral malaria: cohort description and baseline results. Am J Trop Med Hyg. 2017;97:225–31. PubMed PubMed Central Google Scholar
- Holding PA, Stevenson J, Peshu N, Marsh K. Cognitive sequelae of severe malaria with impaired consciousness. Trans R Soc Trop Med Hyg. 1999;93:529–34. CAS PubMed Google Scholar
- Boivin MJ, Gladstone MJ, Vokhiwa M, Birbeck GL, Magen JG, Page C, et al. Developmental outcomes in Malawian children with retinopathy-positive cerebral malaria. Trop Med Int Health. 2011;16:263–71. PubMed Google Scholar
- Idro R, Kakooza-Mwesige A, Asea B, Ssebyala K, Bangirana P, Opoka RO, et al. Cerebral malaria is associated with long-term mental health disorders: a cross sectional survey of a long-term cohort. Malar J. 2016;15:184. PubMed PubMed Central Google Scholar
- Ssenkusu JM, Hodges JS, Opoka RO, Idro R, Shapiro E, John CC, et al. Long-term behavioral problems in children with severe malaria. Pediatrics. 2016;138:e20161965. PubMed PubMed Central Google Scholar
- Idro R, Kakooza-Mwesige A, Balyejjussa S, Mirembe G, Mugasha C, Tugumisirize J, et al. Severe neurological sequelae and behaviour problems after cerebral malaria in Ugandan children. BMC Res Notes. 2010;3:104. PubMed PubMed Central Google Scholar
- Postels DG, Birbeck GL. Children with retinopathy-negative cerebral malaria: a pathophysiologic puzzle. Pediatr Infect Dis J. 2011;30:953–6. PubMed PubMed Central Google Scholar
- Ngoungou EB, Koko J, Druet-Cabanac M, et al. Cerebral malaria and sequelar epilepsy: first matched case-control study in Gabon. Epilepsia. 2006;47:2147–53. PubMed Google Scholar
- Christensen SS, Eslick GD. Cerebral malaria as a risk factor for the development of epilepsy and other long-term neurological conditions: a meta-analysis. Trans R Soc Trop Med Hyg. 2015;109:233–8. PubMed Google Scholar
- Opoka RO, Bangirana P, Boivin MJ, John CC, Byarugaba J. Seizure activity and neurological sequelae in Ugandan children who have survived an episode of cerebral malaria. Afr Health Sci. 2009;9:75–81. PubMed PubMed Central Google Scholar
- Ibiwoye MO, Howard CV, Sibbons P, Hasan M, van Velzen D. Cerebral malaria in the rhesus monkey (Macaca mulatta): observations on host pathology. J Comp Pathol. 1993;108:303–10. CAS PubMed Google Scholar
- Pasini EM, Zeeman AM, van der Wel A, Kocken CHM. Plasmodium knowlesi: a relevant, versatile experimental malaria model. Parasitology. 2018;145:56–70. PubMed Google Scholar
- Rest JR. Cerebral malaria in inbred mice I A new model and its pathology. Trans R Soc Trop Med Hyg. 1982;76:410–5. CAS PubMed Google Scholar
- Hunt NH, Golenser J, Chan-Ling T, Parekh S, Rae C, Potter S, et al. Immunopathogenesis of cerebral malaria. Int J Parasitol. 2006;36:569–82. CAS PubMed Google Scholar
- Desruisseaux MS, Gulinello M, Smith DN, Lee SC, Tsuji M, Weiss LM, et al. Cognitive dysfunction in mice infected with Plasmodium berghei strain ANKA. J Infect Dis. 2008;197:1621–7. PubMed PubMed Central Google Scholar
- White NJ, Turner GD, Medana IM, Dondorp AM, Day NP. The murine cerebral malaria phenomenon. Trends Parasitol. 2010;26:11–5. PubMed PubMed Central Google Scholar
- Riley EM, Couper KN, Helmby H, Hafalla JC, de Souza JB, Langhorne J, et al. Neuropathogenesis of human and murine malaria. Trends Parasitol. 2010;26:277–8. PubMed Google Scholar
- Craig AG, Grau GE, Janse C, Kazura JW, Milner D, Barnwell JW, et al. The role of animal models for research on severe malaria. PLoS Pathog. 2012;8:e1002401. CAS PubMed PubMed Central Google Scholar
- de Souza JB, Hafalla JC, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology. 2010;137:755–72. PubMed Google Scholar
- Langhorne J, Buffet P, Galinski M, Good M, Harty J, Leroy D, et al. The relevance of non-human primate and rodent malaria models for humans. Malar J. 2011;10:23. PubMed PubMed Central Google Scholar
- Ghazanfari N, Mueller SN, Heath WR. Cerebral malaria in mouse and man. Front Immunol. 2018;9:2016. PubMed PubMed Central Google Scholar
- Renia L, Grau GE, Wassmer SC. CD8+T cells and human cerebral malaria: a shifting episteme. J Clin Invest. 2020;130:1109–11. CAS PubMed Google Scholar
- de Miranda AS, Lacerda-Queiroz N, de Carvalho Vilela M, Rodrigues DH, Rachid MA, Quevedo J, et al. Anxiety-like behavior and proinflammatory cytokine levels in the brain of C57BL/6 mice infected with Plasmodium berghei (strain ANKA). Neurosci Lett. 2011;491:202–6. PubMed Google Scholar
- Brant F, Miranda AS, Esper L, Gualdron-Lopez M, Cisalpino D, de Souza DDG, et al. Suppressor of cytokine signaling 2 modulates the immune response profile and development of experimental cerebral malaria. Brain Behav Immun. 2016;54:73–85. CAS PubMed Google Scholar
- Campos AC, Brant F, Miranda AS, Machado FS, Teixeira AL. Cannabidiol increases survival and promotes rescue of cognitive function in a murine model of cerebral malaria. Neuroscience. 2015;289:166–80. CAS PubMed Google Scholar
- Liu M, Solomon W, Cespedes JC, Wilson NO, Ford B, Stiles JK. Neuregulin-1 attenuates experimental cerebral malaria (ECM) pathogenesis by regulating ErbB4/AKT/STAT3 signaling. J Neuroinflammation. 2018;15:104. PubMed PubMed Central Google Scholar
- D’Orleans-Juste P, Akide Ndunge OB, Desbiens L, Tanowitz HB, Desruisseaux MS. Endothelins in inflammatory neurological diseases. Pharmacol Ther. 2019;194:145–60. PubMed Google Scholar
- Dai M, Freeman B, Bruno FP, Shikani HJ, Tanowitz HB, Weiss LM, et al. The novel ETA receptor antagonist HJP-272 prevents cerebral microvascular hemorrhage in cerebral malaria and synergistically improves survival in combination with an artemisinin derivative. Life Sci. 2012;91:687–92. CAS PubMed PubMed Central Google Scholar
- Freeman BD, Martins YC, Akide-Ndunge OB, Bruno FP, Wang H, Tanowitz HB, et al. Endothelin-1 mediates brain microvascular dysfunction leading to long-term cognitive impairment in a model of experimental cerebral malaria. PLoS Pathog. 2016;12:e1005477. PubMed PubMed Central Google Scholar
- Darling TK, Mimche PN, Bray C, Umaru B, Brady LM, Stone C, et al. EphA2 contributes to disruption of the blood-brain barrier in cerebral malaria. PLoS Pathog. 2020;16:e1008261. CAS PubMed PubMed Central Google Scholar
- Varo R, Crowley VM, Sitoe A, Madrid L, Serghides L, Kain KC, et al. Adjunctive therapy for severe malaria: a review and critical appraisal. Malar J. 2018;17:47. PubMed PubMed Central Google Scholar
- Hemmer CJ, Kern P, Holst FG, Nawroth PP, Dietrich M. Neither heparin nor acetylsalicylic acid influence the clinical course in human Plasmodium falciparum malaria: a prospective randomized study. Am J Trop Med Hyg. 1991;45:608–12. CAS PubMed Google Scholar
- Charunwatthana P, Abul Faiz M, Ruangveerayut R, Maude RJ, Rahman MR, Roberts LJ, et al. N-acetylcysteine as adjunctive treatment in severe malaria: a randomized, double-blinded placebo-controlled clinical trial. Crit Care Med. 2009;37:516–22. CAS PubMed PubMed Central Google Scholar
- Maude RJ, Silamut K, Plewes K, Charunwatthana P, Ho M, Abul Faiz M, et al. Randomized controlled trial of levamisole hydrochloride as adjunctive therapy in severe falciparum malaria with high parasitemia. J Infect Dis. 2014;209:120–9. CAS PubMed Google Scholar
- Watt G, Jongsakul K, Ruangvirayuth R. A pilot study of N-acetylcysteine as adjunctive therapy for severe malaria. QJM. 2002;95:285–90. CAS PubMed Google Scholar
- Treeprasertsuk S, Krudsood S, Tosukhowong T, Maek-A-Nantawat W, Vannaphan S, Saengnetswang T, et al. N-acetylcysteine in severe falciparum malaria in Thailand. Southeast Asian J Trop Med Public Health. 2003;34:37–42. CAS PubMed PubMed Central Google Scholar
- Warrell DA, Looareesuwan S, Warrell MJ, Kasemsarn P, Intaraprasert R, Bunnag D, et al. Dexamethasone proves deleterious in cerebral malaria A double-blind trial in 100 comatose patients. N Engl J Med. 1982;306:313–9. CAS PubMed Google Scholar
- Namutangula B, Ndeezi G, Byarugaba JS, Tumwine JK. Mannitol as adjunct therapy for childhood cerebral malaria in Uganda: a randomized clinical trial. Malar J. 2007;6:138. PubMed PubMed Central Google Scholar
- van Hensbroek MB, Palmer A, Onyiorah E, Schneider G, Jaffar S, Dolan G, et al. The effect of a monoclonal antibody to tumor necrosis factor on survival from childhood cerebral malaria. J Infect Dis. 1996;174:1091–7. PubMed Google Scholar
- Das BK, Mishra S, Padhi PK, Manish R, Tripathy R, Sahoo PK, et al. Pentoxifylline adjunct improves prognosis of human cerebral malaria in adults. Trop Med Int Health. 2003;8:680–4. CAS PubMed Google Scholar
- Di Perri G, Di Perri IG, Monteiro GB, Bonora S, Hennig C, Cassatella M, et al. Pentoxifylline as a supportive agent in the treatment of cerebral malaria in children. J Infect Dis. 1995;171:1317–22. PubMed Google Scholar
- Hemmer CJ, Hort G, Chiwakata CB, Seitz R, Egbring R, Gaus W, et al. Supportive pentoxifylline in falciparum malaria: no effect on tumor necrosis factor alpha levels or clinical outcome: A prospective, randomized, placebo-controlled study. Am J Trop Med Hyg. 1997;56:397–403. CAS PubMed Google Scholar
- Wenisch C, Looareesuwan S, Wilairatana P, Parschalk B, Vannapann S, Wanaratana V, et al. Effect of pentoxifylline on cytokine patterns in the therapy of complicated plasmodium falciparum malaria. Am J Trop Med Hyg. 1998;58:343–7. CAS PubMed Google Scholar
- Lell B, Kohler C, Wamola B, Olola CH, Kivaya E, Kokwaro G, et al. Pentoxifylline as an adjunct therapy in children with cerebral malaria. Malar J. 2010;9:368. CAS PubMed PubMed Central Google Scholar
- Iosif RE, Ekdahl CT, Ahlenius H, Pronk CJ, Bonde S, Kokaia Z, et al. Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J Neurosci. 2006;26:9703–12. CAS PubMed PubMed Central Google Scholar
- Clelland CD, Choi M, Romberg C, Clemenson GD Jr, Fragniere A, Tyers P, et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–3. CAS PubMed PubMed Central Google Scholar
- Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–42. CAS PubMed PubMed Central Google Scholar
- Gimenez F, de Lagerie S, Fernandez C, Pino P, Mazier D. Tumor necrosis factor alpha in the pathogenesis of cerebral malaria. Cell Mol Life Sci. 2003;60:1623–35. CAS PubMed Google Scholar
- Lou J, Gasche Y, Zheng L, Critico B, Monso-Hinard C, Juillard P, et al. Differential reactivity of brain microvascular endothelial cells to TNF reflects the genetic susceptibility to cerebral malaria. Eur J Immunol. 1998;28:3989–4000. CAS PubMed Google Scholar
- Armah HB, Wilson NO, Sarfo BY, Powell MD, Bond VC, Anderson W, et al. Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J. 2007;6:147. PubMed PubMed Central Google Scholar
- Solomon W, Wilson NO, Anderson L, Pitts S, Patrickson J, Liu M, et al. Neuregulin-1 attenuates mortality associated with experimental cerebral malaria. J Neuroinflammation. 2014;11:9. PubMed PubMed Central Google Scholar
- Patel SN, Berghout J, Lovegrove FE, Ayi K, Conroy A, Serghides L, et al. C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J Exp Med. 2008;205:1133–43. CAS PubMed PubMed Central Google Scholar
- Serghides L, Kim H, Lu Z, Kain DC, Miller C, Francis RC, et al. Inhaled nitric oxide reduces endothelial activation and parasite accumulation in the brain, and enhances survival in experimental cerebral malaria. PLoS ONE. 2011;6:e27714. CAS PubMed PubMed Central Google Scholar
- Gramaglia I, Sobolewski P, Meays D, Contreras R, Nolan JP, Frangos JA, et al. Low nitric oxide bioavailability contributes to the genesis of experimental cerebral malaria. Nat Med. 2006;12:1417–22. CAS PubMed Google Scholar
- Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, McNeil YR, et al. Impaired nitric oxide bioavailability and l-arginine reversible endothelial dysfunction in adults with falciparum malaria. J Exp Med. 2007;204:2693–704. CAS PubMed PubMed Central Google Scholar
- Gramaglia I, Velez J, Chang YS, Caparros-Wanderley W, Combes V, Grau G, et al. Citrulline protects mice from experimental cerebral malaria by ameliorating hypoargininemia, urea cycle changes and vascular leak. PLoS ONE. 2019;14:e0213428. PubMed PubMed Central Google Scholar
- Serghides L, Patel SN, Ayi K, Lu Z, Gowda DC, Liles WC, et al. Rosiglitazone modulates the innate immune response to Plasmodium falciparum infection and improves outcome in experimental cerebral malaria. J Infect Dis. 2009;199:1536–45. CAS PubMed PubMed Central Google Scholar
- Serghides L, McDonald CR, Lu Z, Friedel M, Cui C, Ho KT, et al. PPARgamma agonists improve survival and neurocognitive outcomes in experimental cerebral malaria and induce neuroprotective pathways in human malaria. PLoS Pathog. 2014;10:e1003980. PubMed PubMed Central Google Scholar
- Wilson NO, Solomon W, Anderson L, Patrickson J, Pitts S, Bond V, et al. Pharmacologic inhibition of CXCL10 in combination with anti-malarial therapy eliminates mortality associated with murine model of cerebral malaria. PLoS ONE. 2013;8:e60898. CAS PubMed PubMed Central Google Scholar
- Taoufiq Z, Pino P, Ndilimabaka N, Arrouss I, Assi S, Soubrier F, et al. Atorvastatin prevents Plasmodium falciparum cytoadherence and endothelial damage. Malar J. 2011;10:52. CAS PubMed PubMed Central Google Scholar
- Wiese L, Hempel C, Penkowa M, Kirkby N, Kurtzhals JA. Recombinant human erythropoietin increases survival and reduces neuronal apoptosis in a murine model of cerebral malaria. Malar J. 2008;7:3. PubMed PubMed Central Google Scholar
- Du Y, Chen G, Zhang X, Yu C, Cao Y, Cui L. Artesunate and erythropoietin synergistically improve the outcome of experimental cerebral malaria. Int Immunopharmacol. 2017;48:219–30. CAS PubMed Google Scholar
- Shabani E, Opoka RO, Idro R, Schmidt R, Park GS, Bangirana P, et al. High plasma erythropoietin levels are associated with prolonged coma duration and increased mortality in children with cerebral malaria. Clin Infect Dis. 2015;60:27–35. CAS PubMed Google Scholar